Location: Home >> Detail
Immunometabolism. 2020;2(3):e200024. https://doi.org/10.20900/immunometab20200024
 ,
Taher E. Taher 1,2,
Rizgar A. Mageed 1,*
,
Taher E. Taher 1,2,
Rizgar A. Mageed 1,*
1 Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Queen Mary University of London, London EC1M 6BQ, UK
2 Institute of Immunology and Immunotherapy, College of Medical & Dental Sciences, University of Birmingham, Birmingham B15 2TT, UK
* Correspondence: Rizgar A. Mageed, Jonas Bystrom.
This article belongs to the Virtual Special Issue "Immunometabolism in Autoimmune Diseases"
Lymphocytes mount protective immunity from infectious pathogens and from cancer. Their engagement by target antigens prompt activation, differentiation to effector cells and proliferation. These responses require energy that is generated by cellular metabolic processes, such as glycolysis and oxidative phosphorylation. Glycolysis occurs in the cytoplasm while oxidative phosphorylation occurs in the mitochondria. Energy in the form of adenosine triphosphate is generated from the uptake of glucose, amino acids and free fatty acids. Important recent evidence indicates that naïve and activated lymphocytes and functionally distinct subsets preferentially use different metabolic pathways for their energy needs. Thus, effector Th17 cells primarily use glycolysis to generate energy required for their activities. Memory T and B cells and regulatory T cells, in contrast, rely on mitochondrial metabolism for their energy requirements. Naïve T and B are in a quiescent state with small mitochondria. Extrinsic factors such as oxygen tension and intrinsic substrates can also influence the choice of metabolic pathways and functional flexibility. Studies of lymphocytes in disease states reveal alterations to choices of metabolic pathways from those in lymphocytes in healthy individuals. This article provides an overview of metabolic pathways required for energy generation during homeostasis and those induced during cellular differentiation and responses. Furthermore, we explore available evidence for altered metabolic pathway induction in some autoimmune diseases. A focus of the overview will be on helper T lymphocytes involved in chronic inflammation and those that regulate the immune response. The report alludes to the potential that targeting metabolic pathways could provide a strategy for the treatment of chronic diseases.
2-HG: 2-hydroxyglutarate; aa: amino acid; Acetyl-CoA: Acetyl Coenzyme A; ACC: acetyl-CoA carboxylase; AID: Activation-induced cytosine deaminase; AhR: Aryl-hydrocarbon receptor; ALDA: Aldolase A; AMP: Adenosine monophosphate; AMPK: Adenosine monophosphate-Activated kinase; Atg7: Autophagy related 7; ATP: adenosine triphosphate; BAFFR: B cell receptor; BCR: B cell receptor; dNTP: deoxynucleoside triphosphate; Drp1: dynamin-related protein 1; FADH2: Flavin adenine dinucleotide; FAO: Fatty acid oxidation; GC: Germinal center; GLUT1: Glucose transporter 1; GSK3: Glycogen synthase kinase 3; GTP: Guanidine triphosphate; HIF-1α: Hypoxia induced factor 1α; HK: Hexokinase; NAD+/NADH: Nicotinamide adenine dinucleotide; mTOR: Mammalian target of rapamycin; OXPHOS: oxidative phosphorylation; PDH: Pyruvate dehydrogenase; PDP2: PDH phosphatase subunit 2; PDK1: Pyruvate dehydrogenase kinase; PFK: Phosphofructokinase; PI3-K: Phosphoinositide-3-OH kinase; PK: Pyruvate kinase; PKCβ: Protein kinase Cβ; PP2A: Protein Phosphatase 2A; PPP: Pentose phosphate pathway; Raptor: Rapamycin-associated TOR protein; Rictor: Rapamycin-insensitive companion of mTor; ROS: reactive oxygen species; SHM: Somatic hyper mutations; Sirt1: Sirtuin-1; STIM1: Stromal interaction molecule 1; TCA: tricarboxylic acid cycle; TCR: T cell receptor; VHL: von Hippel-Lindau
The differentiation and activation of T and B lymphocytes are regulated by differential metabolic activities. Energy sources are taken up to generate adenosine-tri-phosphate (ATP), which is then used in all energy-requiring cellular enzymatic activities. All metabolic activities are divided and compartmentalized between rapid glycolysis in the cytoplasm and slow metabolism in the mitochondria-residing tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS). Recent studies indicate that intrinsic requirements of different lymphocyte subsets determine which metabolic pathway predominates as a source of their energy generation needs. Furthermore, there appears to be regulatory processes that support such preferences [1,2]. Dysregulation of metabolic pathways, or their regulators, can either promote or suppress manifestations of chronic diseases.
This article provides an overview of metabolic processes that underpin T and B lymphocyte activation and differentiation into functional subsets. Of the T cell subsets, the focus will be on Th17 cells that provide protective immunity but can also be involved in autoimmune and chronic inflammatory diseases. We will explore how dysregulation of cellular metabolism can potentially relate to two chronic autoimmune diseases. The role of glycolysis, TCA cycle (also known as the citric acid and Krebs cycle) and the respiratory chain, involved in OXPHOS and how these pathways are interlinked will be explored.
Glycolysis is a basic metabolic pathway that generates ATP through the metabolism of carbon sources such as glucose. Glucose is actively imported into the cytoplasm using membrane localized glucose transporters (GLUTs) where it is phosphorylated. Ten enzymes are involved in this pathway, with the end product being two molecules of pyruvate and four molecules of ATP. There are 3 enzymes with regulatory functions: hexokinase (HK), phosphofructokinase (PFK) and pyruvate kinase (PK). These 3 enzymes promote irreversible changes to their metabolites (Figure 1).
During glycolysis, the co-factor nicotinamide adenine dinucleotide (NAD+) is used to generate NADH. NAD+ has to be re-oxidised for glycolysis. Anaerobic conversion of pyruvate to lactate generates NAD+ in a fast and pH-lowering reaction. This reaction can also take place when oxygen is available, as is the case for glycolysis in tumour cells and T lymphocytes (the Warburg effect). NADH can be re-oxidised in the mitochondria but this requires transportation through membranes. This process reduces Flavin adenine dinucleotide (FAD) to FADH2, which is a cofactor in OXPHOS [3].
Pyruvate can also be used for energy generation when it is transported into the mitochondria and converted into acetyl-CoA. This requires the conversion of NAD+ to NADH, and then citrate in the first step of the TCA cycle. Citrate can be exported out again and converted to cytoplasmic acetyl-CoA and oxaloacetate. Acetyl-CoA is carboxylated by acetyl-CoA carboxylase 1 (ACC1) into malonyl-CoA, a precursor for phospholipid and cholesterol generation. In Th17 cells, phospholipids are generated from citrate derived from glutamate as pyruvate is not transported into the mitochondria [4].
The anabolic pentose phosphate pathway (PPP) in the cytoplasm is required for generating NADPH and ribose 5-phosphate. NADPH is required for fatty acid generation due to its reducing capacity and for protection from radical oxygen species (ROS). Ribose 5-phospate, in contrast, is a precursor required for nucleotide and amino acid production. Glucose-6-phosphate, generated by HK is the substrate for the PPP [3].
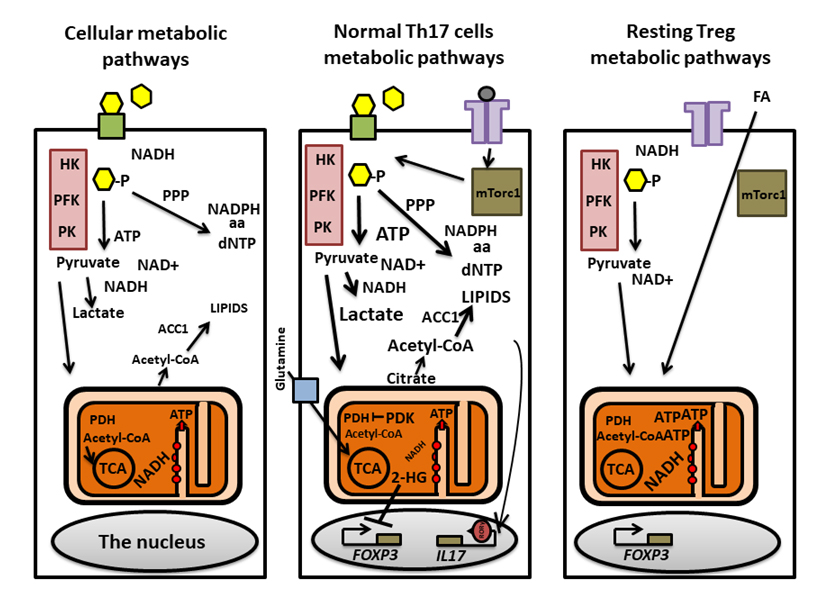 Figure 1. A general overview of metabolic pathways and pathways selectively induced in Th17 and Treg lymphocytes. Basic metabolic pathways involved in generating ATP are shown in the left figure. Glucose (yellow hexagons) enters the cytoplasm through GLUT1 (green boxes) and is metabolized by the glycolysis pathway (purple boxes). HK, PFK and PK modify substrates irreversibly generating pyruvate and NAD+. Pyruvate can be converted to lactate to re-generate NADH for further glycolysis. Metabolites derived from glucose can also be used in the PPP which generates precursors for aa,dNTP and NADPH. Pyruvate entering the mitochondria is used as a substrate for the TCA cycle. PDH converts pyruvate to acetyl-CoA that is, subsequently, metabolized to generate NADH and FADH2 (the latter not shown). TThe electron transport chain (red circles inside the mitochondria) is located in cristae in the mitochondrial inner membranes. NADH and FADH2 are used in OXPHOS to generate ATP. Acetyl-CoA, is converted to citrate and exported from the mitochondria, is then carboxylated byACC1 and used for lipid synthesis. Th17 cell metabolism (middle figure) involves engagement of the TCR and subsequent activation of the glycolysis promoting mTORc1. Mitochondrial import of pyruvate is restricted due to high level of the PDH-deactivating PDK. ACC1 utilizes citrate and generate precursors for lipid synthesis in Th17 cells. Sterols generated from lipid precursors are ligands augmenting RORγt in the cells. The TCA cycle and OXPHOS rely on imports of glutamine (blue box; glutamine transporter SLC1A5), which is metabolized to generate the TCA cycle metabolite α-ketoglutarate. During this process, 2-HG is generated, which in turn can inhibit transcription of the FOXP3 gene, thereby, preventing transdifferentiation of Th17 cells to Tregs. The metabolism of resting Tregs is distinct from Th17 cells’ (right). Tregs rely on the uptake of external fatty acids (FA) and FAO for OXPHOS dependent ATP generation and for the generation of aa and dNTP precursors.
Figure 1. A general overview of metabolic pathways and pathways selectively induced in Th17 and Treg lymphocytes. Basic metabolic pathways involved in generating ATP are shown in the left figure. Glucose (yellow hexagons) enters the cytoplasm through GLUT1 (green boxes) and is metabolized by the glycolysis pathway (purple boxes). HK, PFK and PK modify substrates irreversibly generating pyruvate and NAD+. Pyruvate can be converted to lactate to re-generate NADH for further glycolysis. Metabolites derived from glucose can also be used in the PPP which generates precursors for aa,dNTP and NADPH. Pyruvate entering the mitochondria is used as a substrate for the TCA cycle. PDH converts pyruvate to acetyl-CoA that is, subsequently, metabolized to generate NADH and FADH2 (the latter not shown). TThe electron transport chain (red circles inside the mitochondria) is located in cristae in the mitochondrial inner membranes. NADH and FADH2 are used in OXPHOS to generate ATP. Acetyl-CoA, is converted to citrate and exported from the mitochondria, is then carboxylated byACC1 and used for lipid synthesis. Th17 cell metabolism (middle figure) involves engagement of the TCR and subsequent activation of the glycolysis promoting mTORc1. Mitochondrial import of pyruvate is restricted due to high level of the PDH-deactivating PDK. ACC1 utilizes citrate and generate precursors for lipid synthesis in Th17 cells. Sterols generated from lipid precursors are ligands augmenting RORγt in the cells. The TCA cycle and OXPHOS rely on imports of glutamine (blue box; glutamine transporter SLC1A5), which is metabolized to generate the TCA cycle metabolite α-ketoglutarate. During this process, 2-HG is generated, which in turn can inhibit transcription of the FOXP3 gene, thereby, preventing transdifferentiation of Th17 cells to Tregs. The metabolism of resting Tregs is distinct from Th17 cells’ (right). Tregs rely on the uptake of external fatty acids (FA) and FAO for OXPHOS dependent ATP generation and for the generation of aa and dNTP precursors.
The TCA cycle is required for oxidative metabolism in the mitochondria. The cycle also generates substrates for other compartments. There are 8 enzymes in the TCA cycle that utilise acetyl-CoA to generate three NADH, one FADH2 and one guanidine triphosphate (GTP) molecule. For this process, pyruvate is transported into mitochondria to be converted to acetyl-CoA by pyruvate dehydrogenase (PDH). In the first step, the acetyl-group from acetyl-CoA is transferred to oxaloacetate to form citrate. NADH and FADH2 are subsequently consumed by the respiratory chain to generate ATP. FADH2 binds to succinate dehydrogenase and this complex becomes part of both the citric acid cycle and OXPHOS. The enzyme transfers electrons to coenzyme Q. Although new acetyl-CoA molecules are constantly supplied to the TCA reaction, other metabolites are re-formed for each cycle. If required, pyruvate can be used to form oxaloacetate, instead of acetyl-CoA, by pyruvate carboxylase. The increasing amounts of oxaloacetate generated, in turn, increases the capacity of the TCA cycle, thus, allowing for more metabolism of acetyl-CoA and energy generation. Furthermore, imported glutamate can act as substrate for the TCA cycle [5]. Mitochondria are adaptable and can respond to the increasing energy production by structural rearrangements and an increase in size [2,6]. Interestingly, unconstrained mitochondrial growth is reported to be associated with autoimmune disease [7,8].
There is a stringent regulatory process that prevents the overproduction of NADH and ATP. Excessive amounts of NADH inhibit 4 enzymes in the TCA cycle including PDH. Acetyl-CoA inhibits PDH while citrate inhibits the glycolytic enzyme PFK. Calcium ions (Ca2+) can reach high levels in the mitochondria and this activates pyruvate dehydrogenase phosphatase (PDP) that activates PDH. Dysregulation of PDP is also reported to be associated with autoimmune diseases [9].
Oxidative PhosphorylationOXPHOS is induced in the inner membrane folds of the mitochondria, the cristae [10,11]. The pathway involves electron transport (respiratory chain) with succinate and NADH from the TCA cycle as substrates. The constant oxidation and transfer of protons to the intermembrane space generates a membrane potential during the process of OXPHOS in what is called “the proton motive force”. Four enzyme complexes transport electrons from oxidised compounds, guided by endogenous iron-sulphur cores, to eventually reduce either ubiquinol (Complexes 1 and 2), cytochrome C (Complex 3) or oxygen (Complex 4); in the last case generating water as a by-product. Protons are transferred to the intermembrane space due to the gradient created by the continuous electron transfer [10]. Due to the increase in positive charges in the intermembrane space, protons return to the mitochondrial matrix. This transfer is an active process where protons pass through the fifth complex of OXPHOS, the ATP synthetase. ATP is generated in response to the revolving motion of the protein complex’s head when protons are returning to the mitochondrial matrix but this is dependent on the NADH/NAD+ ratio thereby influencing the proton motion force and membrane potential. OXPHOS also generates ROS, which is as well regulated by the proton motive force. ROS generated by electron transport complexes 1 and 3, are required for basic cellular processes and redox sensitive transcription factors but increased production can cause damage to the cell. One way to control effects of ROS is by the production of radical scavengers such as glutathione. The exact information of how ROS is generated is lacking but increased NADH/NAD+ ratio, possibly the abundance of succinate and local oxygen tension are likely factors involved [12].
The Mammalian Target of Rapamycin (mTOR) and Adenosine Monophosphate-Activated Kinase (AMPK)mTOR is a cytoplasmic sensor of metabolite availability and the level of ROS. The mTOR complex is a serine/threonine kinase that regulates glycolysis, protein synthesis, cell proliferation, motility and autophagy. mTOR exists in different forms, mTORC1, which contains subunit rapamycin-associated TOR protein (Raptor), and mTORC2, which contains subunit rapamycin-insensitive companion of mTOR (Rictor). mTORC1 is a large protein complex located in the endosome in lymphocytes and other cells when nutrients are available. Leucine and Arginine are the most important amino acids for its activation but mTORC1 requires all 20 amino acids ([13,14], Figure 1). AMPK, a cytoplasmic sensor of stress, negatively regulates mTOR. It functions by detecting the availability of ATP in relation to adenosine monophosphate (AMP); low ATP to AMP ratio activates AMPK leading to fatty acid oxidation and the inhibition of fatty acid and protein synthesis. AMPK also increases OXPHOS and mitochondrial gene translation to enzymes in the TCA cycle and the respiratory chain [15].
Naïve T lymphocytes differentiate into one of a number of effector subsets, Th1, Th2, Th17 and Treg cells. Th1 cells provide immunity to intracellular bacteria and viruses. Th2 cells are involved in immunity to extracellular bacteria and parasites. Th17 cells play pivotal role in immunity to fungal and extracellular bacterial infections. Tregs, in contrast, inhibit the emergence of autoreactive effector lymphocytes and suppress inflammation. When naïve T lymphocytes are activated, their differentiation to effector subsets is determined by the type and functional status of antigen presenting cells and by cytokines. For example, IFNγ, IL-12 and TNFα prompt naïve T lymphocytes to differentiate to Th1 cells. IL-4 and IL-5 promote naïve T cells to differentiate to Th2 cells while IL-23, IL-1β and IL-6 promote differentiation to Th17 cells. Tregs can be programmed in the thymus to become natural Tregs. In the periphery, the differentiation of naïve T lymphocytes to Tregs is facilitated by TGFβ. Recent studies have revealed that Th17 cells manifest a plasticity in their functional nature prompted by environmental ques. Th17 cells are, for example, found in the intestinal mucosa to provide immunity and mucosal membrane repairs. They can also produce IL-10 due to stimulation by commensal bacteria. Effector Th17 cells can also transdifferentiate to other T helper subsets upon immune response needs and the cytokine milieu. Thus, Th17 cells can transdifferentiate to Th1-like cells (ex-Th17) or to Tregs [16,17]. Such transdifferentiation is manifested by differential activation of selective metabolic pathways [18]. It has been reported that Th17 to Treg transdifferentiation is dependent on the aryl-hydrocarbon receptor (AhR) which is induced by the tryptophan metabolite kynurenine [17]. Depending on the environment, kynurenine can increase IL-17 production by the Th17 cells but, perhaps paradoxically, can also augment Tregs. In this respect, it is interesting to note that polarized differentiation to Th17 cells and Tregs is associated with the advent of autoimmune diseases and cancer, respectively [16,19,20]. Th17 cells is a focus the T cell section of the review because of their association with diseases and their metabolic requirements particularly in relation to their transdifferentiation to Tregs.
T lymphocytes that leave the thymus to populate the periphery, display a quiescent phenotype and use the OXPHOS metabolic pathway for their energy needs. When activated, these T lymphocytes can upregulate mTORC1, which induces glycolysis that is associated with differentiation of the cells to Th17 cells (in a Th17 polarizing environment). In contrast, inhibition of mTOR promotes naïve T lymphocytes to differentiate to Tregs ([21,22] Figure 1). The requirement for glycolysis in Th17 cell differentiation is highlighted by the fact that the inhibitor of pyruvate conversion to Acetyl-CoA in mitochondria, pyruvate dehydrogenase kinase (PDK1), is expressed at high levels but not in other T cell subsets. When acetyl-CoA generation is inhibited, pyruvate is metabolized to lactate in the cytosol (Warburg’s effect). In this setting, Glut1 transports glucose into cells and is upregulated during cellular proliferation [23]. Therefore, inhibition of PDK selectively suppresses Th17 cell development but enhances the expression of FoxP3 that promotes Treg generation [1]. Studies in which Th17 cells and Tregs were treated with PDK inhibitors revealed an increase in ROS levels in both cell subsets due to increased OXPHOS. Tregs express the ROS scavenger glutathione and thioredoxin-1 and can, therefore, handle oxidative stress better than Th17 cells. This may explain why Th17 cells express and are reliant on PDK expression [1]. Some of the glucose enter the PPP pathway for rapid generation of ribose-5-phosphate as precursors for amino acid and nucleotide synthesis [3]. As influx of pyruvate into the mitochondria is blocked in Th17 cells, the cells rely on imported glutamine that is converted to glutamate to provide a substrate for the TCA cycle as ROS from OXPHOS is required for basic functions of effector Th17 cells [24,25]. One product of glutamate, L-(S)-2-hydroxyglutarate (2-HG) is, therefore, overproduced in Th17 cells, compared with Tregs. 2-HG promotes methylation of the Foxp3 promoter and, thereby, inhibits the development of inducible Tregs [5]. Imported glutamine also facilitates leucine uptake through the transporter protein LAT, thereby, increasing activation of mTORC1. Glutaminolysis of imported glutamine further increases levels of glutathione for protection of the vulnerable Th17 cells from ROS [26].
Acetyl-CoA generated via citrate can be exported into the cytosol. This acetyl-CoA is metabolised by ACC1 to generate precursors for fatty acid synthesis and phospholipid generation in Th1, Th2 and Th17 cells but not Tregs, which are instead reliant of FAO [3,4,27]. Furthermore, inhibition of ACC1 during Th17 cell differentiation induces FOXP3 expression and Tregs differentiation [4]. The importance of ACC1 for Th17 cells is highlighted by the fact that sterols generated during fatty acid synthesis act as ligands to augment expression of the Th17 cell-specific transcription factor RORγt [28]. In this context, it is noteworthy that Th17 cells differentiate better than any other T cell subset during cell stress. Th17 cells, thus, differentiate better at low oxygen levels and in high NaCl concentration settings [29,30]. The ability to differentiate to Th17 cells during cell stress is highlighted in studies of the protein ATF4. This protein is upregulated and, in turn, regulates gene transcription during metabolite restrictions. It induces both glycolysis and the TCA cycle and maintains OXPHOS and the redox state [31]. Indeed, Atf4-deficient CD4+ T cells have defects in redox homeostasis, proliferation, differentiation, and cytokine production. Furthermore, T cells deficient in ATF4 develop to Th17 cells during stress [32]. Th17 cells can also develop into a pathogenic IFN-γ-producing “ex-Th17” phenotype cells and contribute to autoimmune disease pathogenesis under stress and metabolic insufficiency [16]. Two recent studies have assessed the role of metabolism in the transdifferentiation of Th17 cells to IFNγ-producing pathogenic cells. In one study, the requirement for mTORC1 was identified. Thus, T cells deficient in the mTORC1 subunit Raptor developed a stem cell-like character. These cells were driven by the transcription factor Tcf-1 associated with stem cells and with low anabolic activity. Th17 cells with intact mTORC1 can undergo transdifferentiation and will express Tbet-1 and IFNγ. These transdifferentiated ex-Th17 cells showed high metabolic activity and synthesised cholesterol [18]. In the second study, induced overexpression of STAT3 in Th17 cells resulted in increased cellular uptake of Ca2+ through TCR-induced stromal interaction molecule 1 (STIM1). The influx of Ca2+ was required to promote pathogenic Th17 cells’ transdifferentiation but these cells, paradoxically, did not produce IFNγ. The pathogenicity of these cells was, apparently, dependent on mitochondrial OXPHOS and integrity of their cristae to prevent Th17 cells from the release of harmful ROS [1,32].
When the response of effector cells is achieved, a small population of inactive memory cells are retained. The retention of memory T cells, as naïve T cells, is reliant on OXPHOS and fatty acid uptake [33]. However, the cells will have larger mitochondria and more enzymes involved in glycolysis in their cytoplasm than naïve T cells such that memory T cells are promptly ready for any new challenges [33].
According to some estimates, up to 5–10% of T cells in the periphery are regulatory T cells [34]. These cells rely on the TCA cycle and OXPHOS more than other effector cells. Tregs have higher levels of proteins of the respiratory chain than Th17 cell subsets [1]. Ablation of complex 3 in the respiratory chain specifically in Tregs showed that the complex was dispensable for their survival but crucial for their suppressive effects. Complex 3 deficiency in Tregs also resulted in increased levels of 2-HG in mitochondria. A non-functional respiratory chain is known to result in increased NAD/NADH ratio and the conversion of α-ketoglutarate to 2-HG by lactate- or malate-dehydrogenase. It is not known, however, whether 2-HG in this setting silences the FoxP3 promoter [35]. Furthermore, Tregs do not synthesise fatty acids but rely on uptake from exogenous sources [4]. However, when Tregs are stimulated by their target antigen, the cells manifest increased reliance on glycolysis that is dependent on mTOR activity [36].
Aerobic glycolysis and mTOR activation are, therefore, important for the expansion of Th17 cells and their transdifferentiation to ex-Th17 cells. Several explanations have been suggested for the process of favouring the low-efficiency glycolysis during proliferation. Amino acid precursor generation using the PPP in proliferating Th17 helper cells can prevent the inhibitory activities of enzymes from the glycolytic pathway [37]. Rapid phospholipid production for the generation of membranes diverts metabolites away from the TCA cycle [4]. Furthermore, glycolysis may prevent excessive generation of Th17 cells and harmful ROS through the electron transport chain [1]. Resting Tregs, in contrast, are reliant on the TCA cycle, oxidation of fatty acids and OXPHOS. Usage of mice deficient in various metabolic components have also revealed that changes of the metabolic state of cells can facilitate transdifferentiation of Th17 cells to Tregs and vice versa [1,5].
B lymphocytesRelative to T lymphocyte metabolism, there is significantly less information on B lymphocyte metabolism. Predictably, however, metabolic demands vary during B cell differentiation with the highest being during the early stages their generation and proliferation in the bone marrow and during antigen-driven responses. In addition, B cells that express the CD5 protein (B1 cells) are bio-energetically more active than regular CD5 negative B2 cells. Thus, B1 cells have been shown to have higher rates of glycolysis and OXPHOS than B2 cells [38].
In the circulation, naive B lymphocytes are maintained and primed for growth and proliferation by continual tonic signalling through the B cell receptor (BCR) and the B cell activating factor receptor (BAFFR). Activation through both pathways employs the phosphoinositide-3-OH kinase (PI3-K) [39–41]. The main metabolic sequels of signalling through the BCR and BAFFR in this setting appears to be the preservation of homeostatic mitochondrial functions and harnessing anabolism [42]. BCR-mediated signalling activates protein kinase Cβ (PKCβ), which enhances the glycolytic flux [43]. Interestingly, PKCβ is also downstream effector of PI3-K, which enhances PKC activity through PDK1 and mTOR complex 2 (mTORC2) [44]. Activity of mTORC1 is, however, regulated through sensing the concentration of nutrients such as amino acid and the presence of oxygen and this feeds back to inhibit PI3K [45]. This suggests the existence of a cross-talk between nutrient-sensing and kinase signalling resulting in bidirectional regulation. Additionally, mTORC1 activity is crucial for B cell differentiation and antibody production [46,47]. Paradoxically, however, mTORC1 can be inhibited by active AMP-activated kinases (AMPK), which has been shown to link nutrients and energy status with different cell functions. This, however, appears to have no impact on antibody production [48]. Thus, although active AMPK inhibits mTORC1, through several mechanisms, primary antibody production is not affected in AMPKa1-null mice [49]. In the absence of antigen, or growth-inducing factors, constitutively active glycogen synthase kinase-3 (GSK3) acts as a metabolic rheostat in resting B cells, supporting cell survival through restricting protein synthesis and reducing cell size [50] (Figure 2).
Both naïve and memory B cells share a quiescent feature but with important distinctions. Thus, memory B cells have the ability to persist in the absence of BAFF [51]. Additionally, studies of autophagy related 7 (Atg7) gene-deficient mice indicate that in contrast to naïve B cells autophagy has a crucial role in the persistence of memory B cells [52,53]. Of further note is that results from Atg5 loss-of-function studies indicate that another component of the autophagosome contributed to homeostasis and primary humoral responses [54,55]. Additionally, a number of studies have revealed that immunoglobulin isotype expression could differentially impact persistence and survival of memory B cells. Thus, IgM expression contributes to persistence and survival of memory B cell more than IgG [56,57]. This suggests that BCR (auto) specificity, and/or specific intrinsic signalling may be features of particular Ig isotype [58–62]. However, the impact of expression of different BCR isotypes on B cell metabolism, nutrient requirements and anabolic activities are yet to be revealed.
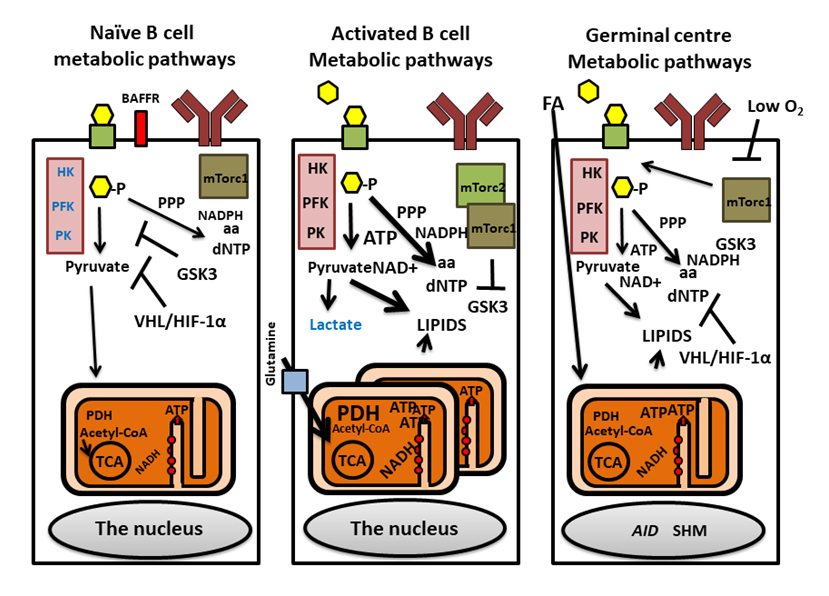 Figure 2. Metabolic pathways regulating naïve, activated and germinal center B cells. Naïve B cells manifest low metabolic activity, low glycolysis (purple boxes with enzymes, HK, PFK and PK), small mitochondria and low OXPHOS. GSK3, regulates this low metabolic status. The VHL protein regulates metabolism and survival via HIF1α. Activated B cells (middle) utilize glucose (yellow hexagons), via PPP to generate of dNTP and lipids but with little Lactate produced. During activation, the mitochondria remodel to many punctate small mitochondria with OXPHOS activation that is driven by amino acids such as glutamate (Glutamine transporter SLC1A5, blue box) [2]. mTORC2 regulates mTORC1 promoting glycolysis by suppressing GSK3. During germinal center reactions, B cells proliferate rapidly in the dark zone, acquiring SHMs mediated by AID which is driven by glycolysis and OXPHOS (right figure). In the light zone, and during subsequent B cell selection, oxygen level is low and metabolic activity is regulated by GSK3. This environment activates VHL while inhibiting mTORc1 and glycolysis. B cells rely on OXPHOS using FA derived from membranes of surrounding dying B cells. GLUT1: green boxes with black borders; BCR: indicated as brown Y-shaped antibodies; BAFFR: red box.
Figure 2. Metabolic pathways regulating naïve, activated and germinal center B cells. Naïve B cells manifest low metabolic activity, low glycolysis (purple boxes with enzymes, HK, PFK and PK), small mitochondria and low OXPHOS. GSK3, regulates this low metabolic status. The VHL protein regulates metabolism and survival via HIF1α. Activated B cells (middle) utilize glucose (yellow hexagons), via PPP to generate of dNTP and lipids but with little Lactate produced. During activation, the mitochondria remodel to many punctate small mitochondria with OXPHOS activation that is driven by amino acids such as glutamate (Glutamine transporter SLC1A5, blue box) [2]. mTORC2 regulates mTORC1 promoting glycolysis by suppressing GSK3. During germinal center reactions, B cells proliferate rapidly in the dark zone, acquiring SHMs mediated by AID which is driven by glycolysis and OXPHOS (right figure). In the light zone, and during subsequent B cell selection, oxygen level is low and metabolic activity is regulated by GSK3. This environment activates VHL while inhibiting mTORc1 and glycolysis. B cells rely on OXPHOS using FA derived from membranes of surrounding dying B cells. GLUT1: green boxes with black borders; BCR: indicated as brown Y-shaped antibodies; BAFFR: red box.
From the information cited above, there is a clear need for better understanding of the metabolic requirements for the activation of antigen-specific B cells and the metabolic reprogramming such processes entail. Several metabolic adaptations and reprogramming are likely to be induced because of signalling initiated by the engagement of the BCR and/or other co-receptors. For example, there is evidence that glucose uptake increases dramatically, at least partially, through upregulation of glucose transporter 1 (GLUT1). However, this could also be due to increased downstream demands (i.e., utilization of glucose-6-phosphate). Noteworthy is that IL-4 receptor engagement, or the engagement of the BCR or TLR4 enhances glucose oxidation and promotes pyruvate generation [63,64]. Interestingly, diminished GLUT1-mediated glucose uptake is characteristic of B cell anergy and metabolically quiescent state [65]. Thus, manipulating the glucose uptake pathway either through enhancing or down regulating GLUT-1 could provide a useful therapeutic avenue for manipulating peripheral tolerance mechanisms [66,67]. Furthermore, B cell activation dramatically enhances the uptake of specific amino acids, including glutamine through upregulating the expression of solute transporter proteins such as the accessory chain CD98hc (Slc3a2). The uptake of specific amino acids is essential for robust B cell proliferation [68], and is rapidly upregulated in PDK1- and AKT-dependent manner [69]. The increases in the concentration of specific amino acids helps maintain an optimal ability to have signal flux through the mTORC1 [70].
The maintenance of plasma cells and antibody production are mainly regulated through the expression of transcription factors Blimp-1 and IRF4 [71]. Intriguingly, loss of Blimp-1 expression in plasma cells severely affects the expression of several amino acid transporters and mTORC1 activity [71]. Furthermore, IRF4 plays a key role in B cell commitment to the germinal centre response and differentiation to plasma cells [72]. Although the direct implication of IRF4 in regulating B cell metabolic reprogramming is still to be established, in T cells IRF4 regulates the expression of genes that enhance glucose uptake and promote the glycolytic response, while exerting no effects on oxidative phosphorylation [73]. Of further interest, are the recent findings that long-lived plasma cells employ greater mitochondrial respiration and have greater mitochondrial respiratory reserve capacity than short-lived plasma cells [74]. Additionally, metabolic tracing analyses have revealed that long-lived plasma cells have higher glucose uptake than short-lived plasma cells. However, most of the glucose was devoted to the synthesis of carbohydrates needed for glycosylation of the antibodies to be secreted than feeding the TCA cycle.
With regards to the metabolic status of B cells, recent studies have revealed an important role for hypoxia. Thus, hypoxia was shown to be pronounced in the germinal centres in mice and that this contributes to reduced proliferation, enhanced apoptosis, diminished class switch recombination to IgG2c and reprograming B cell metabolism [46,50,75]. Hypoxia inducible factors (HIF) 1 and 2 promote the glycolytic program through the induction of aldolase A (ALDA), phosphoglycerate kinase 1 (PGK1), and the M2 isoform of pyruvate kinase (PKM2) expression [76]. Moreover, PKM2 also acts as a co-activator of HIF1-α and, therefore, mediates a positive feedback loop that augments the glycolytic program [77]. Consistent with HIF-mediated inhibition of both mTORC1 and c-Myc activity, the anabolic growth and respiration of B cells may be affected [46,78]. Both HIF and c-Myc promote glycolysis, perhaps, predominantly during the proliferative phase in the dark zone [79]. HIF suppresses the TCA cycle and respiration while c-Myc promotes mitochondrial bioenergy [80,81]. However, a recent study has revealed that mitochondrial respiration can and does take place during low oxygen tension. This study concluded that B cell metabolism in the light zone of germinal centres is driven by FAO. It was suggested that FA derived from the membranes of B cells dying due to the lack of co-stimulation provide these FAs ([82], Figure 2). In line with HIF that regulates IRF4 expression, hypoxia promotes the differentiation of B cells to plasma cells [73,75].
As cited above, lymphocytes responses are regulated through metabolic homeostasis. In healthy individuals, lymphocytes undergo metabolic reprogramming during immune responses to infectious pathogens or to self-proteins. Such metabolic reprogramming enables lymphocytes to generate the energy required to provide immunity/anergy. The reprogramming changes anabolic and catabolic pathways to support heightened energy demands. Thus, lymphocytes have a metabolic flexibility that enables them use multiple sources for energy generation. Defects in the magnitude of energy generation and flexibility, either becoming hyper-metabolic or hypo-metabolic leads to the skewing of lymphocyte metabolism towards augmented or reduced fuel consumption and biosynthesis, respectively. Increases in fuel availability undermines immune regulation in favour of reduced tolerance leading, potentially, to autoimmune diseases. A hypo-metabolic state, by contrast, results in a reduction in lymphocyte metabolic flexibility and defective immunity. Thus, defective metabolism can critically perturb lymphocyte homeostasis and a loss of self-tolerance [83]. During activation, lymphocytes adapt to temporary increases in energy demands. However, in various disease states, dysregulated signalling in lymphocytes causes constitutive activation of kinases, potentially through chronic triggering of autoreactive lymphocytes and energy stress. Thus far, the most focus is on the metabolic balance of T lymphocytes and their contribution to autoimmune diseases.
Rheumatoid Arthritis (RA)Rheumatoid arthritis (RA) is an autoimmune inflammatory disease characterised by immune mediated damage to joints [84]. Although a number of studies have assessed metabolic properties of T cells in RA, few studies have carried out similar studies in B cells. Environmental and genetic factors combine to drive an immune response involving T lymphocytes and inflammatory cytokine production to cause pathology [19]. A recent study reported identifying a defect affecting patients’ T cell metabolism in RA. RA T cells were reported to contain reduced levels of one of the regulators of glycolysis, 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase (PFKFB). This enzyme regulates levels of a glucose metabolite, fructose 2,6-bisphosphate which, in turn, is a known activator of the glycolysis enzyme PFK. Therefore, patients experience reduced expression of PFK, with the consequence being that its substrate, glucose-6-phosphate, is directed to the PPP instead of being further metabolized by glycolysis. Over-activity of PPP, in turn, leads to a reducing intracellular status, excessive NADPH generation and reduced mitochondrial ROS generation [85]. This reductive state leads to increased ageing of T cells and increased production of GM-CSF, IFNγ and IL-17. Such defects in T cells increases their propensity for migration to tissues [86,87]. In addition, excessive levels of lactate in the inflamed synovium has been shown to induce OXPHOS-driven Th17 cell differentiation and their retention in the synovium [88]. Intriguingly, studies in RA patients and in animal models of arthritis have revealed that Th17 cells play key pathological roles especially in patients whose disease is, apparently, not primarily driven by TNFα [89,90]. It is currently not known what drives the expansion of Th17 cells in RA. However, it is likely that the synovial microenvironment promotes hyper-metabolism, which alters the dynamics of T lymphocyte differentiation in favour of Th17 cells. In contrast, there are a number of studies that have examined whether Tregs have lost the ability to supress self-reactivity in RA [91–93]. Assessments of the metabolic status of Tregs in RA is currently lacking. However, although the TCA cycle and OXPHOS are functional, it is likely that the inflammatory response and lipid biogenesis reported are associated with detrimental metabolic pathways for proper functionality of the cells [1,27]. Different therapies are under consideration to augment Tregs as potential treatment of RA and autoimmune diseases in general. Enhancement of TNFR2 activation has been shown to promote the proliferation and the restoration of the effector functions of Tregs [94]. In addition, there is evidence that low doses of IL-2 promote Treg activation in RA [95,96]. In vitro expansion of Tregs as a therapeutic approach for treating patients with RA is another option under consideration. Both rapamycin, used to inhibit mTOR activation, and TNFR2 agonists have been used in such studies [34,97].
Systemic Lupus Erythematosus (SLE)SLE is a systemic autoimmune disease associated with circulating autoantibodies to nuclear antigens, the formation of immune complexes and increased complement activation leading to reduced levels of key complement components. There is also evidence for reduced activation of the first complement component which causes a defect in the uptake and removal of apoptotic cell debris [98]. In addition, there is evidence for abnormalities in the T and B lymphocyte compartments. For T lymphocytes, for example, there is evidence for increased Th17 cell numbers, reduced IL-2 production by T cells and reduced Treg cell numbers and their suppressive activities [99]. There is also evidence for dysregulated metabolism in Th17. For example, SLE Th17 cells manifest high levels of glycolysis and mTORC1 activation. The mTOR inhibitor Sirolimus (rapamycin) was shown in a clinical trial to inhibit Th17 cell proliferation and to increase Treg numbers. This treatment was suggested to obviate the need for steroid therapy in treated patients [100]. T cells from patients with SLE also exhibit increased mitochondrial mass and dysfunction, characterized by elevated mitochondrial transmembrane potential. In lupus-prone mice, combination therapy with inhibitors of glycolysis (2-deoxy-D-glucose) and OXPHOS (metformin) restored T cell metabolism and ameliorated disease [67]. In addition to enhanced mTORC activation, T cells in patients with SLE are characterized by large dysfunctional mitochondria. A recent study highlighted that the expression of a regulator, or endocytic recycling, Rab4A, is increased, apparently driven by oxidative stress [8]. Increased endocytic recycling of surface proteins such as the CD4, CD3zeta and the endosome-bound Drp1 leads to their increased degradation and reduced membrane levels. Reduced availability of Drp1 leads to increased mitochondrial size in T cells in SLE patients [7]. Interestingly, the gene that encodes Rab4A has been associated with susceptibility to SLE [8]. In a murine model of lupus, increased mTORC1 activation in liver cells prior to disease was shown to result in reduced Drp1 expression and dysfunctional OXPHOS. One component of the electron transport chain, complex I, was overexpressed in the liver of these mice. The production of phosphatidylethanolamines and phosphatidylserine is regulated in mitochondrial membranes. Interestingly, dysfunctional mitochondria in the liver of these mice lead to the production of anti-phospholipid autoantibodies with the mice developing anti-phospholipid syndrome and both were reduced by treatment with rapamycin [8].
A metabolomics study of plasma from lupus patients revealed high activity of the PPP pathway. The study also identified increased levels of kynurenine. Whether these findings relate to the increase in Th17 cells in SLE patients is, however, unknown. Nevertheless, treatment with N-acetylcysteine (a therapy given to improve another aspect of lupus pathology; glutathione deficiency) resulted in reduced kynurenine levels in the patients [101] (Figure 3).
The protein phosphatase 2A (PP2A), which regulates Th17 cell differentiation and activation, is increased in lupus T cells. PP2A regulates the production of IL-2, expression of CD3zeta-chain of the TCR and remodels DNA to enhance IL17 production [102,103]. The increase in PP2A in lupus T cells causes aberrant TCR signalling and cell functions. Interestingly, however, suppression of the Pp2a gene in Tregs resulted in increased mTOR activity which was associated with the development of disease [104,105]. In addition, the enzyme PDH, which converts pyruvate to acetyl-CoA, is activated by PDH phosphatase subunit 2 (PDP2). In line with the notion that Th17 cells are primarily driven by glycolysis, expression of PDP2 is reduced in Th17 cells in SLE. In a mouse model of lupus, overexpression of PDP2 in CD4+ T cells diminished Th17 cell activity. In this setting, inducible cAMP early repressor, part of inducible cAMP early repressor/cAMP response element modulator a factor known to regulate Th17 cells in lupus, was found binding and regulating the Pdp2 promoter [9]. Evidence for how subtle changes in levels of metabolic co-factors can cause epigenetic alterations that, in turn, regulate cellular phenotype has come from studies of T cell expressing CD38. This membrane receptor is an activation marker and a NAD+ nucleosidase generating ADP-ribose and nicotinamide. The role of CD38’s nucleosidase in cell functions was highlighted in studies where Th1/Th17 cells with low levels of CD38 were shown to have increased NAD+ levels and increased capacity to eradicate tumor cells. The enhanced anti-tumor property of these Th1/17 cells was dependent on the increased NAD+-dependent activity of the histone deacetylase Sirtuin-1 (Sirt1) [106]. In lupus disease, however, the number of CD38+CD8+ T cells is increased in a subgroup of patients that succumb to infections. Increased degradation of NAD+ in these cells was shown to inhibit Sirt1. Inhibition of this DNA deacetylase resulted in activation of the histone methylase and transcriptional repressor Enhancer of zeste homolog 2. Activity of CD38 in CD8+ T cells from patients with lupus, thus, resulted in reduced IFNγ production compared with CD38−CD8+ T cells, perhaps, explaining the succumbing of these patients to infections [107].
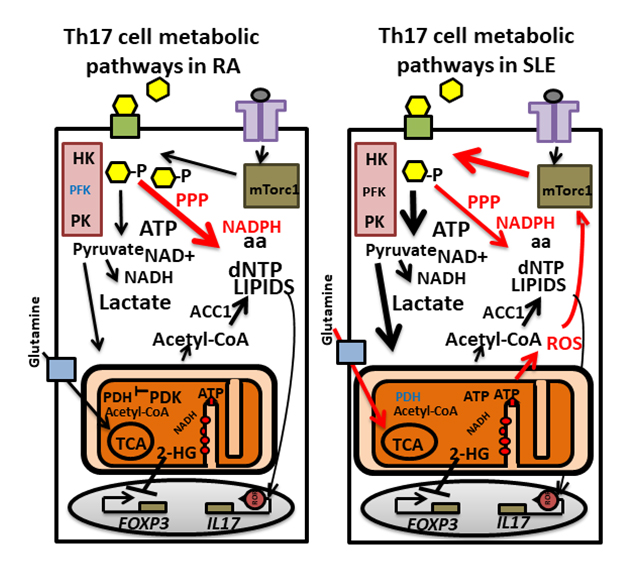 Figure 3. Altered metabolic pathways in Th17 cells from patients with rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). Main metabolic pathways are altered in Th17 cells in RA and SLE compared with healthy individuals (red arrows). In RA Th17 cells, PFKFB is upregulated resulting in reduced activity of glycolysis (purple box with enzymes HK, PFK and PK) enzymes PFK (blue color). Excessive amounts of glucose (yellow hexagons) are, therefore, shunted through the PPP rendering the cells to be in a reductive state with high levels of NADPH. The cells develop an inflammatory senescence phenotype leading to high level production of IL-17, TNFα and IFNγ. Th17 cells in SLE display increasd glycolysis and OXPHOS and enlarged mitochondria. Signaling through mTORC1 (green box) induces glycolysis and increases PPP output. Increased ROS generated during OXPHOS (electron transport by complexes (red small circles)) further activates mTORC1. PDH is suppressed in SLE Th17 cells and the TCA cycle relies on glutamine import (glutamine transporter SLC1A5: blue box). GLUT1: green boxes on the membrane with black borders; TCR: violet T shaped boxes on the membrane.
Figure 3. Altered metabolic pathways in Th17 cells from patients with rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). Main metabolic pathways are altered in Th17 cells in RA and SLE compared with healthy individuals (red arrows). In RA Th17 cells, PFKFB is upregulated resulting in reduced activity of glycolysis (purple box with enzymes HK, PFK and PK) enzymes PFK (blue color). Excessive amounts of glucose (yellow hexagons) are, therefore, shunted through the PPP rendering the cells to be in a reductive state with high levels of NADPH. The cells develop an inflammatory senescence phenotype leading to high level production of IL-17, TNFα and IFNγ. Th17 cells in SLE display increasd glycolysis and OXPHOS and enlarged mitochondria. Signaling through mTORC1 (green box) induces glycolysis and increases PPP output. Increased ROS generated during OXPHOS (electron transport by complexes (red small circles)) further activates mTORC1. PDH is suppressed in SLE Th17 cells and the TCA cycle relies on glutamine import (glutamine transporter SLC1A5: blue box). GLUT1: green boxes on the membrane with black borders; TCR: violet T shaped boxes on the membrane.
These observations suggest that differential induction of metabolic pathways in T cells could promote the expansion/activation of lupus-promoting T cells and disease.
For B lymphocytes, high levels of mTOR activation were reported to be associated with disease activity [108]. In the Roquin mouse model of lupus, activation of AMPK and inhibition of mTOR in B cells, reduced their differentiation to germinal centre (GC) and plasma cells and correlated with disease activity [109]. Metformin, which is an activator of AMPK, was shown to be beneficial in treating SLE patients as well as reducing disease activity in mouse models of lupus [67,110,111].
Overall, lymphocytes in patients and lupus-prone mice display high glycolysis, increased oxygen consumption and dysregulated mitochondrial growth [67,112,113]. Indeed, over-expression of Glut1, the glucose transporter, in mice led to the accumulation of activated CD4 T cells, increased numbers of germinal centres B cells, production of autoantibodies and immune complex deposition in kidneys [114]. Additionally, treatment with 2-deoxy-D-glucose (2DG), a glycolysis inhibitor, reduced T and B cell metabolism and prevented the development of disease while its combination with metformin, reversed lupus pathogenesis and disease severity in mice [67,115]. Targeting glycolysis may, thus, provide a therapeutic strategy for treating lupus patients [116].
Recent studies of lymphocyte metabolism have revealed selective association of specific metabolic pathways with distinct functional T and B lymphocytes subsets. Intricate selection of dominant metabolic pathways determines whether T lymphocytes differentiate to pro-inflammatory or regulatory cells. In B lymphocytes, there are also distinctive prominence of specific metabolic pathways in resting naïve, memory and plasma cells. The use of genetically engineered mice with cell specific deficiencies in selective metabolic enzymes has identified mediators important for the maintenance of specific lymphocyte subsets. These studies and studies using human lymphocyte subsets have revealed the importance of mediators such as acetyl-CoA, 2-HG and Ca2+ and enzymes in OXPHOS in the differentiation of functional subsets. Furthermore, there is evidence for altered activation of metabolic pathways in lymphocytes from patients with a range of diseases including autoimmune diseases compared with healthy controls. Interestingly, however, altered induction of metabolic pathways appears to differ in different diseases and to occur through different mechanisms. Thus, T cells in RA patients show defects in the regulation of the glycolytic pathway whilst, T cells in SLE patients have overactive glycolysis and mTOR activity, which seem to promote the expansion of Th17 cells.
JB: Designed, reviewed, collected information and wrote on basic metabolic pathways and involvement in T lymphocytes in health and disease; TET: Designed, reviewed, collected and wrote on metabolic pathways in B lymphocytes in health and disease; RAM: Designed the review and overviewed all the gathered data by JB and TET on metabolic pathways in lymphocytes in health and in autoimmune diseases, helped write and edit the text, incorporated the references and finalised the text.
The authors declare no conflicts of interest.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
Bystrom J, Taher TE, Mageed RA. Metabolic Pathways Underpinning Lymphocyte Differentiation and Responses in Health and Disease. Immunometabolism. 2020;2(3):e200024. https://doi.org/10.20900/immunometab20200024

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




