Location: Home >> Detail
Immunometabolism. 2019;1:e190011. https://doi.org/10.20900/immunometab20190011
 ,
Lucien Platon 1,2,3,
Carmen Buchrieser 1,2,**
,
Lucien Platon 1,2,3,
Carmen Buchrieser 1,2,**
1 Institut Pasteur, Unité de Biologie des Bactéries Intracellulaires, 75015 Paris, France
2 CNRS-UMR 3525, 75015 Paris, France
3 Faculté des Sciences, Université de Montpellier, 34095 Montpellier, France
* Correspondence: Pedro Escoll, Tel.: +33-0-1-44-38-9540; Carmen Buchrieser, Tel.: +33-0-1-45-68-8372.
This article belongs to the Virtual Special Issue "Immunometabolism and Inflammation"
Beyond oxidative phosphorylation (OXPHOS), mitochondria have also immune functions against infection, such as the regulation of cytokine production, the generation of metabolites with antimicrobial proprieties and the regulation of inflammasome-dependent cell death, which seem in turn to be regulated by the metabolic status of the organelle. Although OXPHOS is one of the main metabolic programs altered during infection, the mechanisms by which pathogens impact the mitochondrial electron transport chain (ETC) complexes to alter OXPHOS are not well understood. Similarly, how changes on ETC components affect infection is only starting to be characterized. Herein we summarize and discuss the existing data about the regulation of ETC complexes and super-complexes during infection, in order to shed some light on the mechanisms underlying the regulation of the mitochondrial OXPHOS machinery when intracellular pathogens infect eukaryotic host cells.
OXPHOS, oxidative phosphorylation; ETC, electron transport chain; mROS, mitochondrial reactive oxygen species; UQ, ubiquinone; CytC, cytochrome C; ANT, Adenosine Nucleotide Translocator; UCP, uncoupling proteins; SDH, succinate dehydrogenase; mPTP, mitochondrial permeability transition pore; TLR, toll-like receptor; PRR, pattern recognition receptor; LPS, lipopolysaccharide; HEV, hepatitis E virus; DAMM, Danger Associated Metabolic Modification; mΔψ, mitochondrial membrane potential; RET, reverse electron transport; ECSIT, Evolutionarily Conserved Signaling Intermediate in Toll pathway; TRAF6, TNF receptor associated factor 6; TCA, tricarboxylic acid; NO, nitric oxide; RISP, Rieske Iron Dulfur Protein; CVB3, Coxsackievirus B3; MAVS, mitochondrial antiviral signaling protein
Although perception of mitochondria as signaling hubs that regulate immune responses has exploded in recent years [1–7], how infection modulates mitochondrial functions is not well studied. Beyond oxidative phosphorylation (OXPHOS), mitochondria have immune functions that counteract infection, such as the regulation of cytokine production, the generation of metabolites with antimicrobial proprieties or the regulation of inflammasome-dependent cell death (recently reviewed in [8]). Compelling evidence suggests that the metabolic status of the organelle regulates these additional functions of mitochondria, while immune signaling can also regulate mitochondrial metabolic functions [1,8–10]. Moreover, it seems that metabolic shifts are essential in the activation of adaptive and innate immune cells [11–13], and metabolic processes such as autophagy are used as cell-autonomous defense systems of both immune and somatic cells to degrade invading pathogens during infection, a process called xenophagy [14–16]. In order to exploit during infection the key roles that these organelles play in the host cell, intracellular pathogens have evolved mechanisms to target the host metabolism and specifically mitochondria [17–19]. Moreover, as intracellular pathogens can obtain the energy resources required for their growth only from the infected host cell, any metabolic alteration in the host cell should also impact the pathogen, suggesting that the control of cellular metabolism might be one determinant for the outcome of an infection. Therefore, metabolic interactions are increasingly considered when studying the host-pathogen interface [20].
OXPHOS is one of the main metabolic programs that is known to be modulated during infection [21]. OXPHOS is driven by the orchestrated action of different protein complexes located at mitochondrial cristae and embedded in the inner mitochondrial membrane, that is known as the electron transport chain (ETC)(Figure 1). Although the ETC is composed of five complexes (Complex I to V), the action of additional proteins, such as the ubiquinone (UQ), the Cytochrome C, the Adenine Nucleotide Translocator (ANT), or the uncoupling proteins (UCPs), as well as the formation of super-structures, are required to perform OXPHOS at mitochondria according to cellular needs [22,23](Figures 1 and 2). The structure of mitochondria is determinant for OXPHOS activity at different levels. First, the compartmentalization of mitochondria allows the efficient performance of the mitochondrial bioenergetics functions. Oxidation of acetyl-CoA derived from carbohydrates, fatty acids, and proteins by the tricarboxylic acid (TCA) cycle is performed in the mitochondrial matrix, while the flow of electrons through the ETC occurs at the inner mitochondrial membrane, and the protons pumped by ETC complexes accumulate in the inter-membrane space (Figures 1 and 2). Second, the assembly of ETC complexes into super-complexes seems to influence OXPHOS performance by facilitating proton translocation from the mitochondrial matrix to the intermembrane space (Figure 2), which generates the mitochondrial membrane potential (mΔψ). The largest assembled super-complex that was named the respirasome is constituted of Complex I, two copies of Complex III and up to 4 copies of Complex IV [24](Figure 2). In the presence of Cytochrome C and UQ, the respirasome creates the predominant part of the proton-motive force as a unique block. Thus, the generated mΔψ allows the utilization of this proton-motive force to perform OXPHOS-dependent synthesis of ATP through the activity of the mitochondrial FOF1-ATP synthase, known as the complex V [21,23]. Complex V can oligomerize forming dimers, tetramers or hexamers, which might increase cristae stability [25](Figure 2). Interestingly, accessory proteins such as ANT have been suggested to form a super-complex with the FOF1-ATP synthase, called the ATP synthasome [25]. In contrast, Complex II seems not to be part of any super-complex [26]. Importantly, it has been recently proposed that different pathological conditions, such as neurodegenerative disorders or infection of macrophages with pathogenic bacteria, produce alterations in the composition and activity of ETC complexes and super-complexes [27–29].
In this review, we summarize and discuss the current understanding of the roles that mitochondrial ETC complexes, super-complexes and OXPHOS-related proteins play, when eukaryotic host cells are infected by pathogens (viruses, bacteria or parasites), as well as some of the mechanisms underlying the regulation of mitochondrial OXPHOS machinery during infection.
 Figure 1. The mitochondrial Electron Transport Chain (ETC) comprises complexes I to V (CI to CV) as well as other accessory proteins, such as Ubiquinone (UQ) and Cytochrome C (CytC). These proteins work together to oxidize reduced compounds such as NADH, FADH2 or Succinate and finally transfer electrons to O2, thereby generating water and creating an electrochemical gradient of protons used by Complex V to synthesize ATP (oxidative phosphorylation, OXPHOS). Dotted lines represent the flow of electrons. IMM: inner mitochondrial membrane; IMS: inter-membrane space.
Figure 1. The mitochondrial Electron Transport Chain (ETC) comprises complexes I to V (CI to CV) as well as other accessory proteins, such as Ubiquinone (UQ) and Cytochrome C (CytC). These proteins work together to oxidize reduced compounds such as NADH, FADH2 or Succinate and finally transfer electrons to O2, thereby generating water and creating an electrochemical gradient of protons used by Complex V to synthesize ATP (oxidative phosphorylation, OXPHOS). Dotted lines represent the flow of electrons. IMM: inner mitochondrial membrane; IMS: inter-membrane space.
 Figure 2. The mitochondrial cristae is where the ETC and accessory OXPHOS proteins organize in individual complexes and supercomplexes (SC). These are embedded in the inner mitochondrial membrane (IMM). Complexes and SC pump protons (H+) into the inter-membrane space (IMS) to create the electrochemical gradient used by Complex V to generate cellular energy (lyme colored stars) by synthesizing ATP from ADP. ADP originates from the cytoplasm (not shown), crosses the outer mitochondrial membrane (OMM) and the IMM (not shown), and enters in the matrix by the action of the Adenine Nucleotide Translocator (ANT). The electrochemical gradient can be dispersed by the action of uncoupler proteins (UCP), which generate heat (red colored star).
Figure 2. The mitochondrial cristae is where the ETC and accessory OXPHOS proteins organize in individual complexes and supercomplexes (SC). These are embedded in the inner mitochondrial membrane (IMM). Complexes and SC pump protons (H+) into the inter-membrane space (IMS) to create the electrochemical gradient used by Complex V to generate cellular energy (lyme colored stars) by synthesizing ATP from ADP. ADP originates from the cytoplasm (not shown), crosses the outer mitochondrial membrane (OMM) and the IMM (not shown), and enters in the matrix by the action of the Adenine Nucleotide Translocator (ANT). The electrochemical gradient can be dispersed by the action of uncoupler proteins (UCP), which generate heat (red colored star).
Complex I is the largest complex of the ETC. Complex I is composed of 46 subunits in mammalian cells, where seven of the subunits are encoded by mitochondrial DNA while the rest are encoded by nuclear genes located in different chromosomes. The enzymatic complex oxidizes the NADH generated by the TCA cycle and other sources. Complex I transfers two electrons from NADH to UQ, a lipid soluble electron carrier embedded in the lipid bilayer of the IMM, which is reduced to ubiquinol (UQH2). It is thought that Complex I contributes by 40% to the proton-motive energy driving OXPHOS, although the mechanism by which protons are translocated is not yet well defined.
In mammalian cells, Complex I is one of the major sites of production of mitochondrial reactive oxygen species (mROS), a class of highly reactive molecules that have been showed to provide lifespan proprieties [30,31]. The generation of mROS (superoxide O2•− and hydrogen peroxide H2O2) occurs when flowing electrons exit the ETC prior to the reduction of oxygen to water, thus reacting instead with oxygen to form superoxide, which is quickly dismutated to hydrogen peroxide by superoxide dismutases [32]. Generation of mROS by electron leakage at Complex I is a well-established phenomenon. Electrons can flow in both directions at Complex I depending on the fueling substrates, and in both conditions mROS can be produced by electron leakage [33]. During the forward transport, electrons from NADH flow from Complex I to Complex III (via UQ), and mROS is generated at the flavin mononucleotide (FMN) prosthetic group of the enzyme if the NADH/NAD+ ratio is high or if Complex I is inhibited (for example, by rotenone)[34]. However, in conditions of high mΔψ and high concentrations of succinate, part of the electrons at Complex II now flows through the reverse electron transport. They flow from Complex II to Complex I via UQH2, where NAD+ is reduced to NADH, and higher amounts of mROS are produced by a mechanism that seems to involve the reaction of the partially reduced form of the UQ (UQH•−) with O2, which forms mROS [34,35]. However, the exact mechanisms by which electrons are leaked to generate mROS during forward transport or reverse electron transport (RET) are still a matter of investigation and debate [33,34].
The role of mROS as signaling molecules is well established in the regulation of pathways that are important during infection, such as activation of inflammasomes or activation of autophagy (reviewed in [32]). The inflammasomes are high-molecular-weight protein complexes that are assembled in response to inflammatory stimuli through the activation of their pattern-recognition receptors (PRRs). The main consequence of inflammasome assembly is the activation of Caspase-1, which in turn will produce the mature forms of the pro-inflammatory cytokines interleukin 1β (IL1β) and IL18. It has been shown that one of the inflammasomes named NACHT, LRR and pyrin domain-containing protein 3 (NLRP3), can be activated in response to high concentrations of mROS [32,36–38], highlighting the role of mROS in the onset of inflammatory responses. In addition to inflammasome activation, several other downstream effects of mROS have been described [32]. For example, it seems that a specific pool of proteins is oxidized by mROS, which might account for certain functions of mROS [39]. Mitochondrial membrane lipids and mitochondrial DNA can also be affected by the oxidative damage of mROS, which modulates OXPHOS levels [40]. Moreover, inhibition of Complex I by rotenone triggers autophagy by a mechanism involving mROS production, beclin-1 and class III-PI3K [41]. Finally, it is worth to mention that nitric oxide (NO), a small molecule induced during inflammation, inhibits the activity of Complex I by damaging the iron–sulfur centres, which leads to mROS production [42]. We can thus hypothesize that at least some of the major actions elicited by Complex I during infection might be linked to mROS production. In this regard, it has been shown that mROS signaling has a protective role during infection with bacteria, viruses and parasites [1,32,43,44].
One example of which mechanisms may underlie the production of mROS and Complex I during infection is the activation of the PRRs called toll-like receptors (TLRs) by microbial compounds, such as lipopolysaccharide (LPS), that lead to the translocation of TNF receptor associated factor 6 (TRAF6) to mitochondria, where it interacts with Evolutionarily Conserved Signaling Intermediate in Toll pathway (ECSIT), a protein involved in the assembly of Complex I [45,46]. Consequently, TRAF6-mediated ubiquitination of ECSIT leads to increased mROS production in murine macrophages. Therefore, activation of TLR signaling leads to the modulation of Complex I activity through the action of ECSIT. Interestingly, Salmonella replicates intracellularly to higher numbers within ECSIT-deficient macrophages [46]. As ECSIT is a protein involved in Complex I assembly, it might be possible that bacteria-induced TLR-TRAF6-mediated ECSIT modifications compromise the flow of electrons through the Complex I, which presumably leads to electron leakage and mROS production. However, how mROS contributes then to bacterial clearance was not addressed in this study. Although ROS generated by the NADPH oxidase in the cytoplasm or phagosomes has been shown to posses direct anti-microbial actions, and ROS are exploited as anti-microbial treatments in the clinic [47,48], direct killing of bacteria through mROS (i.e., ROS generated within mitochondria) has not been proven. We can thus speculate that the control of bacterial infection by the TRAF6/ECSIT/mROS axis might be related to the mROS-dependent activation of defense pathways such as the NLRP3 inflammasome or autophagy/xenophagy [32]. Interestingly, it has been recently showed that ECSIT-deficient murine macrophages have a dysfunctional Complex I and rely on a metabolic state that is characterized by reduced OXPHOS and increased glycolysis [49].
GRIM19/NDUFA13 is a Complex I subunit that has also been shown to interact with a PRR, in this case Nucleotide Oligomerization Domain 2 (NOD2), which recognizes bacterial peptidoglycan [50]. GRIM19 seems essential for NF-κB activation downstream of NOD2, and its activity seems protective against Salmonella infection, as silencing GRIM19 in infected host cells increases the numbers of intracellular bacteria [50]. Another study showed that GRIM19 levels and Complex I activity are induced during infection with Staphylococcus saprophyticus, while macrophages deficient for GRIM19 have a reduced ability to kill bacteria and also showed impaired cytokine production [51]. In contrast, for Escherichia coli or Salmonella enterica it has been shown that when infecting murine macrophages, the ETC is reorganized by decreasing Complex I levels and increasing Complex II activity, which impacts also the distribution of Complex I-containing super-complexes at the ETC [28]. Therefore, some bacterial species seem to increase Complex I levels, while other species reduce Complex I levels and exclude it from the ETC.
In addition to bacteria, viruses can also target GRIM19. Human cytomegalovirus (hCMV) encodes a 2.7-kilobase RNA that binds GRIM19 and protects infected cells from apoptotic pathways activated by metabolic stress of Complex I [52]. Avoidance of host cell apoptosis during hCMV infection was due to the metabolic shift from OXPHOS to glycolisis induced by the virus [53], similarly to the metabolic shifts that help cancer cells to avoid cell death [54]. Thus, hCMV-induced inhibition of host cell death can be caused by several mechanisms and not exclusively by the regulation of GRIM19/Complex I.
Finally, recent reports showed the importance of Complex I activity in the production of the pro-inflammatory cytokine IL1β via reverse electron transport -generated mROS, as LPS-activated macrophages decrease OXPHOS-dependent ATP production, increase oxidation of succinate by Complex II, decrease forward transport at Complex I, and increase mΔψ, which all together triggers reverse electron transport and mROS production at Complex I [10]. Reverse electron transport -generated mROS at Complex I in turn activates the transcription of IL1β by an HIF1α-dependent mechanism [10]. Blocking reverse electron transport with Complex I inhibitors (e.g., rotenone or metformin) reduced LPS-induced mROS and IL1β production [10,55]. Moreover, deletion of a Complex I subunit named NDUFS4 showed a profound inflammatory phenotype in mice that includes increased mROS production and metabolic abnormalities [56]. Together, these studies demonstrate that appropriate Complex I functions are necessary to regulate inflammation and, in particular, macrophage metabolism and IL1β production.
In summary, Complex I activity seems to have a protective role during bacterial infection, as deficiency of certain Complex I subunits reduces the clearance of bacteria during infection (Figure 3). Certain of these actions could be mediated by increased mROS at Complex I. Moreover, Complex I levels seem to be decreased during bacterial infection of murine macrophages, which might increase mROS production. Finally, mROS generated at Complex I via reverse electron transport is involved in the production of IL1β, a key pro-inflammatory cytokine to fight infection.
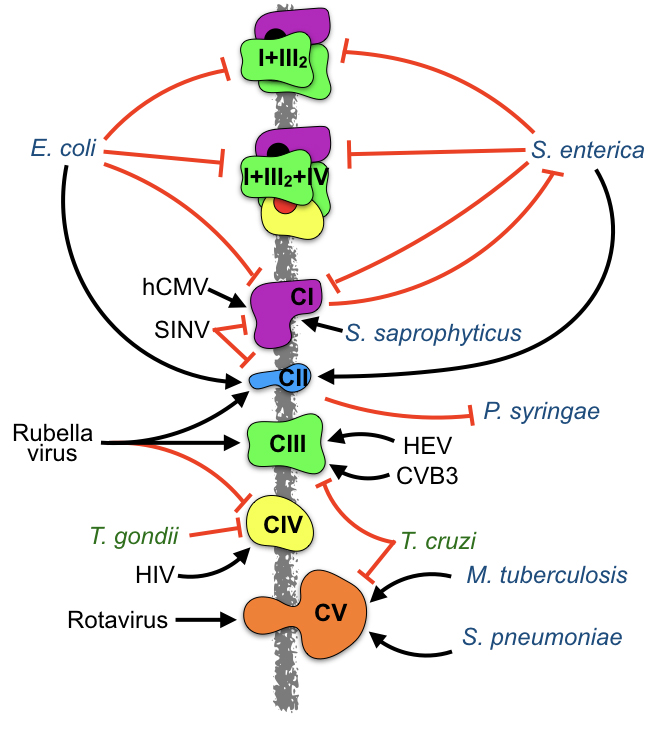 Figure 3. Pathogens influence the levels and activity of ETC complexes and super- complexes. Infection by viruses (black), bacteria (blue), and parasites (green) impact the levels and activity of several ETC complexes. Black arrows: induction. Red lines: inhibition. The influence of pathogens in the formation of super-complexes has been only addressed during E. coli or S. enterica infection of murine macrophages. In addition, ETC complexes also affect infection, as Complex I and Complex II protect from S. enterica and P. syringae infection, respectively.
Figure 3. Pathogens influence the levels and activity of ETC complexes and super- complexes. Infection by viruses (black), bacteria (blue), and parasites (green) impact the levels and activity of several ETC complexes. Black arrows: induction. Red lines: inhibition. The influence of pathogens in the formation of super-complexes has been only addressed during E. coli or S. enterica infection of murine macrophages. In addition, ETC complexes also affect infection, as Complex I and Complex II protect from S. enterica and P. syringae infection, respectively.
Complex II is the smallest complex of the ETC that is comprised of four subunits (SDHA, SDHB, SDHC, SDHD) and represents the second entry point of reducing agents into the ETC. Complex II is the only complex where all subunits are encoded by nuclear DNA, and it is also the only complex that does not pump protons across the inner mitochondrial membrane. In addition to its role in OXPHOS as an ETC component, the complex also participates in the TCA cycle, providing a functional link between these two essential processes [57]. Therefore, Complex II has two intrinsic enzymatic activities, succinate dehydrogenase (SDH activity) and succinate UQ reductase activity (SQR activity). The SDH activity generates FADH2 by the oxidation of succinate to fumarate as part of the TCA cycle, and is performed at the matrix. The SQR activity occurs at the inner mitochondrial membrane where Complex II uses the electrons provided by FADH2 (from the TCA cycle or other sources) to reduce UQ to UQH2, which is further shuffled along the ETC.
As explained before, Complex II has a key role in the production of IL1β via RET/mROS/HIF1α and in antibacterial defense [10,28]. LPS-induced TLR activation of murine bone-marrow derived macrophages (BMDMs) breaks the mitochondrial TCA cycle by a mechanism involving pyruvate kinase M2, leading to increased succinate production and increased Complex II activity [58], which leads to RET and mROS generation at Complex I.
In addition to the aforementioned role of Complex II on RET-derived mROS generation at Complex I, and although it has long been assumed that OXPHOS-derived mROS are only produced at Complex I and Complex III, some reports indicated that Complex II is also a significant direct source of mROS relevant in certain pathological conditions and in cell death [57]. In this regard, a role of complex II as a pH sensor of cell death has been suggested [59]. In this model, a decrease in intracellular pH leads to the specific dissociation of the SDHA/SDHB subunits from membrane-anchored Complex II, thus disappearing from the ETC and accumulating in the matrix. Independent of the ETC, SDH activity of SDHA/SDHB oxidizes succinate to fumarate, while SQR activity cannot be performed, as the downstream acceptors for the electrons (SDHC/SDHD) are missing. In this case, the electron flow is blocked and the electrons are transferred to molecular oxygen instead, generating mROS that can activate apoptosis [60]. However, this mechanism might not operate during bacterial infection, as Complex II seems to remain at the ETC and even increases its activity, while Complex I levels are decreased as mentioned before, upon stimulation of BMDMs with living E. coli or S. enterica [28]. Interestingly, these rearrangements of ETC complexes are absent when BMDM are stimulated with dead bacteria. Increased Complex II activity upon stimulation with living E. coli depend on TLR and NLRP3 signaling, subsequent ROS production by phagosomal NADPH oxidase and the ensuing actions of the ROS-dependent tyrosine kinase Fgr. Moreover, inhibition of Complex II during infection of macrophages with living bacteria led to cytokine levels similar to those of macrophages stimulated with dead bacteria [28]. This study thus identified ETC rearrangements as an early immunometabolic checkpoint that adjusts innate immune responses to bacterial infection, and highlighted the anti-bacterial role of Complex II in murine macrophages.
Complex II activity seems also altered during viral infection in a virus-specific manner. By a non-identified mechanism, the activity of both Complex I and Complex II decreases during infection of a mouse neuroblastoma cell line by Sindbis virus (SINV)[61]. However, in response to Rubella virus infection, a strong increase in complex II activity, a moderate increase of Complex III activity, and a decreased activity of Complex IV was observed in three different human cell lines [62], which seems to indicate that not all viruses increase Complex II activity during infection. Finally, also mitochondrial Complex II is essential for antibacterial and antifungal defense in plants, as plants with a point mutation on Complex II produced less mROS and were more susceptible to infection by the necrotrophic root fungus Rhizoctonia solani as well as by the bacterium Pseudomonas syringae [63].
Therefore, in addition to its role on IL1β production via RET/mROS, Complex II activity seems increased after infection with some viruses and bacteria (Figure 3) and seems essential for antibacterial and antifungal defenses in plants.
Complex III: Cytochrome bc1 ComplexIn human cells Complex III is composed of 10 subunits, which are encoded by both the mitochondrial (cytochrome b) and the nuclear genome (all other subunits). Complex III oxidizes ubiquinol UQH2 and transfers two electrons to two molecules of cytochrome c, a water-soluble electron carrier located within the intermembrane space. During this electron transfer reaction, two protons are taken up from the mitochondrial matrix and four protons are deposited on the other side of the inner mitochondrial membrane. When electron transfer is reduced (by a high membrane potential or Complex III inhibitors such as antimycin A), electrons can leak at complex III and react with molecular oxygen, resulting in the generation of mROS.
Complex III has been shown to be involved in the replication of Hepatitis E virus (HEV). Pharmacological inhibition of Complex III restricted HEV replication at an equivalent level to that observed by ribavirin treatment, a widely used drug for patients with chronic HEV [64]. This restriction of HEV replication seems to involve the mitochondrial permeability transition pore (mPTP), which is a highly evolutionarily conserved, non-selective and regulated pore formed within the inner and outer mitochondrial membrane, permeable to molecules less than 1.5 kDa in size. Its opening connects the mitochondrial matrix and the cellular cytoplasm and results in loss of mΔψ of the proton gradient and of ATP production, which eventually leads to mitochondrial dysfunction and cell death. HEV infection robustly blocked the opening of the mPTP. As inhibition of the mPTP opening with cyclosporin A also attenuated the anti-HEV effect of Complex III inhibitors, the mechanism supporting the antiviral effects of Complex III inhibitors seems to involve the opening of the mPTP. However the mechanism remains unclear as the mPTP composition is still a matter of debate and multiple models have been put forward (reviewed in [65]). For example ETC complexes and OXPHOS-related proteins, such as Complex V or Adenine Nucleotide Translocator [65], have been proposed to be part of the mPTP. Therefore, these results might suggest that during infection HEV blocks mPTP opening, which could delay cell death and support viral replication. In this case, the induction of mPTP opening with Complex III inhibitors would kill the host cells and restrict HEV replication. How Complex III inhibitors induce mPTP opening in the context of HEV infection remains unknown.
Infection of mice with Coxsackievirus B3 (CVB3), which causes myocarditis, revealed that Complex III activity is increased on infected myocardium during viral infection [66]. Increased Complex I and Complex III activity correlated with increased oxidative stress and active cell death. As CVB3 titers were reduced, it was suggested that elevated activities of Complex I and Complex III contribute to mROS generation leading to subsequent mitochondria-related cell death and virus elimination [66]. In contrast, myocardium of mice infected by the parasite Trypanosoma cruzi showed decreased Complex III activity, which compromised respiration and ATP synthesis of infected myocardium [67]. A reduced expression of cytochrome b caused the loss of Complex III activity constituting one mechanism leading to decreased OXPHOS and ATP production in the myocardium of infected mice [67]. Complex III has thus different roles in the myocardium of mice infected by CVB3 or T. cruzi. When mice are infected by the virus, Complex III activity is increased and seems protective, while infection by the parasite decreased Complex III activity and seems deleterious.
For bacterial infections no studies directly investigating the role of Complex III in infected cells are published. However, it was reported that, by using mice with T-cell-specific reduction of the Complex III subunit Rieske Iron Dulfur Protein (RISP), mROS generated at Complex III was required for antigen-specific T cell expansion upon infection with Listeria monocytogenes. RISP was also necessary to generate memory responses [68], highlighting a role of T cells and Complex III in the clearance of L. monocytogenes.
In summary, although Complex III activity seems induced by HEV and CVB3 viruses, it seems protective or deleterious for host cells depending on the virus (Figure 3), but they always seem to involve the regulation of cell death. While Complex III activity is reduced during parasitic infections, its direct role in bacteria-infected host cells remains to be addressed.
Complex IV: Cytochrome c OxidaseComplex IV is composed of 20 subunits in human cells, three of which are encoded by the mitochondrial genome. Complex IV is the last enzyme of the ETC. It receives an electron from each of four cytochrome c molecules, and transfers them to O2, converting it into two molecules of water while four protons are translocated across the inner mitochondrial membrane.
Complex IV activity is regulated during viral infection in a virus- or host cell-specific manner. Rubella virus infection decreased activity of Complex IV in three different human cell lines [62]. However, an increase in Complex IV activity has been described to occur after HIV-1 infection of CD4+ T cell lines, which was associated with viral-induced apoptosis because inhibition of Complex IV by potassium cyanide reduced apoptosis of infected cells [69]. Therefore, the increased Complex IV activity observed might be a specific T cell response to HIV-1 infection. Furthermore, the COX5B subunit of Complex IV was shown to interact with the mitochondrial antiviral signaling protein (MAVS), a mitochondrial outer membrane protein involved in antiviral responses [70,71]. Upon detection of viral genomes by PRRs, MAVS induces the assembly of a signalsome that activates the IRF3- and NF-κB-dependent cytokine production leading to the antiviral immune response [72]. The physical interaction of COX5B with MAVS negatively regulates MAVS-mediated antiviral pathways. While activation of MAVS lead to increased mROS production and COX5B expression, COX5B in turn down-regulated MAVS signaling by reducing mROS production [70]. Importantly, the role of COX5B in the regulation of mROS production was also evidenced in another study [73], thereby highlighting the functions of the COX5B subunit in the regulation of mROS levels, MAVS signaling and the antiviral response [70,71,73].
Measurements of OXPHOS protein levels during Toxoplasma gondii infection showed that the expression of Complex IV is decreased 24 h post-infection [74]. Thus in parasitic infections, Complex IV levels are regulated, what does not seem to be the case during bacterial infection. A role of Complex IV in sepsis, the systemic inflammatory response to infection, has been suggested as myocardial Complex IV is inhibited during sepsis in animal models [75–77]. In these models, oxygen utilization but not delivery seems impaired, a condition referred to as cytopathic hypoxia, suggesting that Complex IV, which is the terminal acceptor of oxygen, might function differently during sepsis [76]. Indeed, septic patients who survived have increased Complex IV activity as compared to non-survivors, independently of the bacterium causing the sepsis [78], which might emphasize the importance of rescuing Complex IV activity to improve survival during sepsis. Importantly, inflammatory signaling triggered by septic conditions leads to the inhibition of Complex IV via tyrosine phosphorylation. Furthermore, similarly to Complex I, NO also inhibits the activity of Complex IV [76,77]. Therefore, for bacterial infections, Complex IV inhibition was addressed during sepsis and are thought to be related to the inflammatory conditions of septic patients and not to a direct regulation of Complex IV by pathogenic bacteria. As rescuing Complex IV activity correlates with a good prognosis, targeting NO-induced Complex IV inhibition may also have a therapeutic interest for sepsis patients.
In summary, Complex IV activity is modulated by viral infection in a virus- or host cell-specific manner, and is decreased during parasitic and bacterial infection (Figure 3), which might be related to infection-associated inflammatory signals.
Complex V: FOF1 ATP SynthaseComplex V is composed of 29 subunits in human cells, which collectively form a rotating molecular machine that consists of two main functional regions, FO and F1, in charge of ATP synthesis. The translocation of protons by Complex I, Complex III and Complex IV creates an electrochemical gradient (a difference in proton concentration across the inner mitochondrial membrane) that fuels the rotation of the motor mechanism of Complex V when protons return to the matrix, allowing the Complex V machinery to synthesize ATP from ADP + Pi. Thus, the mitochondrial FOF1 ATP synthase couples the return of protons located at the intermembrane space by oxidative reactions to the synthesis of ATP, which represents the core of OXPHOS. Besides its ATP synthase activity, Complex V possesses also ATP hydrolase activity, which is supported by an alternative functioning of the motor protein known as the “reverse mode”. This reverse mode of Complex V is induced mainly when proton leakage occurs at the inner mitochondrial membrane or when OXPHOS is compromised in the cell [79].
It has been reported that bacterial infection regulated the levels of diverse Complex V subunits. For example, the protein levels of the ATP5O subunit were markedly enhanced during infection of THP-1 macrophage-like cells with Mycobacterium tuberculosis, while ATP5B and ATP5A1 levels were down-regulated [80]. In the case of murine macrophages infected with Streptococcus pneumoniae, mRNA expression of ATP12A, ATP4A, ATP4B, ATP5J and ATP6V1C2 subunits, as well as ATP5A protein levels, were induced during infection [81]. Interestingly, when macrophages from young and aged mice were compared, infection-associated up-regulation of the Complex V subunit levels only occurred in young macrophages [81], suggesting that aging impacts how Complex V is regulated during infection. However the exact mechanisms of regulation of Complex V subunits during bacterial infection remain to be elucidated.
In the case of viral infections, a report identified ATP5B as having high affinity to the Rotavirus 3’UTR consensus sequence. During Rotavirus infection, ATP5B is bound to the Rotavirus 3’UTR and co-localized with viral RNA and viroplasm, which identifies ATP5B as host factor that supports late-stage virus replication [82]. Another study reported a single amino acid mutation in the cucumber mosaic virus capsid protein that elicits unusual symptoms when plants were infected. This single mutation induced a strong interaction of the virus surface with the F1 region of the Complex V in infected plants, which lethally blocks the rotation of the ATP synthase motor and leads to cell apoptosis and plant death [83]. Therefore, these studies showed that direct interaction of viral proteins with Complex V subunits could regulate the functions of mitochondrial FOF1 ATP synthase and affected viral replication within host cells.
Complex V is also regulated during parasitic infections. Defects on the Complex V activity of cardiac mitochondria appeared in the chronic phase of T. cruzi infection of mice [84], which contribute together with Complex III to the aforementioned decrease of OXPHOS and ATP production in the myocardium of T. cruzi-infected mice [67]. This study suggested a role of ETC complexes and heart mitochondria on the altered cardiovascular functions seen in T. cruzi-infected patients [67]. Another report showed that infection with the yeast-like fungus Pneumocystis carinii induces the over-expression of ATP6 subunit mRNA in the lungs of infected rats [85]. The ATP6 subunit is a key component of the proton channel and one of the two Complex V subunits encoded by the mitochondrial genome. Although the impact of ATP6 mRNA over-expression on the metabolism of host cells during infection was not addressed [85], this study demonstrated that infection by pathogenic fungi altered the expression of Complex V subunits in host cells.
Collectively, these studies show that viral, bacterial, fungal and parasitic infections influence the levels of Complex V subunits and their activity (Figure 3). Whether these changes are induced by the pathogens or are a response of host cells to infection remain to be determined.
Non-ETC Accessory OXPHOS Proteins: Adenine Nucleotide Translocator and Uncoupler ProteinsSome proteins, not strictly belonging to the ETC, have important roles in the regulation of OXPHOS, such as the adenine nucleotide translocator (ANT) and uncoupler proteins (UCP). They are also located at the inner mitochondrial membrane (Figure 2) and seem regulated by infection.
ANT is the most abundant protein in the inner mitochondrial membrane and belongs to the mitochondrial carrier family. It exchanges free ATP with free ADP across the inner mitochondrial membrane. As ADP is the substrate of Complex V, its activity regulating ATP/ADP exchange across the inner mitochondrial membrane also regulates OXPHOS levels [79]. Indeed, the ANT and the FOF1 ATP synthase are not necessarily in directional synchrony, and the conjugation of their forward/reverse directionalities determines mΔψ and OXPHOS levels [79]. Four ANT tissue-specific isoforms exist in humans, ANT1 (SLC25A4), ANT2 (SLC25A5), ANT3 (SLC25A6) and ANT4 (SLC25A31). In addition to their role in ADP/ATP exchange, ANT has been proposed to be also part of the mPTP, thus triggering inner mitochondrial membrane permeabilization and leading irreversibly to cell death [65]. Interestingly, some viral products interact with mitochondrial ANTs, because of their role in apoptosis. Some examples are the porcine circovirus type 2 protein ORF4 that triggers cell death by direct binding to ANT3 [86], the HIV-1 viral protein R that impairs mitochondria axonal transport in primary neuronal cultures by its interaction with ANT [87], the hcmv-miR-UL36-5p miRNA encoded by human cytomegalovirus that inhibits cell death by inhibiting ANT3 expression [88], or the interaction of UL7 with ANT2 in the case of infection by herpes simplex virus 1b [89]. Taken together, these reports suggested that ANT activity is targeted by viruses in order to regulate host cell death.
UCPs are inner mitochondrial membrane proteins that regulate a proton channel that dissipates the electrochemical gradient generated by Complex I, Complex III and Complex IV. However, the energy lost in dissipating the proton gradient via UCPs is not used for biochemical functions, but heat is generated, thus UCP are responsible for thermogenesis. Therefore, UCPs and the ATP synthase, which is also a proton channel, work in parallel at the inner mitochondrial membrane with the electrochemical proton gradient and, although mitochondrial respiration is coupled to ATP synthesis during OXPHOS thanks to the Complex V, this process is finally regulated by UCPs through the controlled dissipation of the gradient. There are five UCP isoforms in mammals, named UCP1 to UCP5, but UCP2 has been the only one linked to infection. It was shown that UCP2-KO mice exhibited increased resistance to microbial infections as compared to wild-type mice. Animals lacking UCP2 were protected against infection with Listeria monocytogenes or T. gondii [44,90]. Interestingly, these studies showed that macrophages from UCP2-KO mice generated more mROS than wild-type mice in response to T. gondii infection but, conversely, another study showed that overexpression of UCP2 reduced mROS production in vitro [91]. Although contradictory, these results suggest a role of UCP2 in the regulation of mROS and thus UCP2 might be a promising target to reduce mROS production in some pathologies [92,93]. In addition, some pathogens seem to induce UCP2 levels. For instance, infection by the bacterium Porphyromonas gingivalis elicited a strong and time-dependent increase in mitochondrial UCP2 levels [94], as well as infection by the parasite Leishmania donovani, which also increases UCP2 activity to suppress mitochondrial oxidative burst and inflammasome activation [95,96].
In summary, pathogens can regulate directly or indirectly accesory OXPHOS proteins such as ANT and UCPs in addition to ETC complexes, which might have an effect on the bioenergetics of the host cells during infection.
The activity of mitochondrial ETC complexes is modulated by viruses, bacteria and parasites during infection of eukaryotic host cells. This mainly leads to bioenergetic alterations in the host cell related to changes in OXPHOS levels and, in many cases, to a regulation of cell death. The link between mitochondrial ETC, OXPHOS levels and cell death remains recurrent not only in infection. One well-known example is CytC, which binds to cardiolipin when it is anchored to the inner mitochondrial membrane and part of the ETC, and it can be released from the mitochondria to initiate apoptosis [65]. Regulation of cell death during infection represents an important strategy for pathogens, which can maintain their niche or kill the host cells depending on their needs. Therefore, the regulation/modulation of ETC activity and OXPHOS levels by pathogens might have not only bioenergetic benefits, but also it might also initiate or delay host cell death.
OXPHOS seems to be a cellular checkpoint. An adequate OXPHOS performance represents normal cellular functions, and an altered, non-adequate OXPHOS performance represents a danger signal for the host cell. We have recently proposed the term DAMM for Danger-Associated Metabolic Modifications [97]. In this model, OXPHOS alteration during infection might be a DAMM for host cells. In this case, the fine-tuning of the ETC machinery and its associated signaling pathways would be key for pathogens to avoid triggering DAMMs and the subsequently elicitied immune defense mechanisms such as inflammasome activation, IL1β production or programmed cell death.
Metabolic shifts from OXPHOS-based metabolic programs to glycolytic-based ones have been observed in multiple cell types and conditions, including immune cells upon activation and infection [4,12,21,54,97]. In the case of infection, the relative contribution of the host and the pathogen to these metabolic shifts remains a matter of debate, as host-driven and pathogen-driven metabolic alterations seem to co-exist during infection [97]. During infection, host cells partially prevent an energetic crisis upon decreased OXPHOS levels by up-regulating glycolytic-based ATP production [76]. On the other hand, pathogens induce metabolic shifts to glycolytic-based programs coupled with biosynthetic pathways to create the conditions allowing their multiplication within infected cells [21]. This debate may be extended to other metabolic shifts seen when OXPHOS is reduced, such as mitochondrial substrate-level phosphorylation (mSLP), a pathway where ATP is generated in the mitochondrial matrix independently of the ETC, mainly by the action of the mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) or the succinate-CoA ligase [98,99]. As it has been shown that mSLP is inhibited upon stimulation of murine macrophages with LPS [100], one might argue that mSPL does not energetically compensate bacterial-induced reduction of OXPHOS due to the blockage by bacterial LPS. However, the finding that patients infected with the hepatitis C virus have up-regulated levels of PEPCK-M might suggest that viral infection induces mSPL [101]. Whether this effect can be seen in other infections, and whether mSLP pathways benefit host cells or pathogens, are interesting questions that remain to be answered.
The study of the OXPHOS machinery during infection has been mainly restricted to the analysis of individual ETC complexes and their subunits. However, although it is well known that ETC complexes assemble at mitochondria in super-complexes during OXPHOS, the investigation of the super-complexes functions and configurations during infection remains scarce. Only one recent report explored this topic by investigating ETC super-complex configuration during infection of murine macrophages with E. coli or S. enterica [28], showing that infection downregulated all Complex I-containing super-complexes.
A very interesting aspect that should be addresed in the future is whether genetic deficiencies of ETC complexes, which are frequent in patients with neurodegenerative, motor and/or cognitive problems, also predispose to infection. Some case reports of patients harboring mitochondrial ETC dieseases include recurrent infections as symptoms, and some reports showed that patients initiated their neurological symtomps after suffering feaver or a well identified infection (Table 1). These clinical cases of patients with deficiencies in ETC complexes might point to a functional link between mitochondrial ETC complexes and infections [102–107].
 Table 1. Genetic deficiencies of ETC complexes seem to be linked to infection.
Table 1. Genetic deficiencies of ETC complexes seem to be linked to infection.
Mitochondrial ETC complexes have been thus proposed as therapeutic targets for human diseases such as autism, cancer or viral infections [64,108,109]. However, their roles regulating host responses to infection or their targeting by pathogens during infections remain largely unknown. Thus the study of ETC complexes during infection will not only bring fundamental knowledge about metabolic host-pathogen interactions, but might also determine whether mitochondrial ETC complexes can be used as targets in the host to tackle infection.
LP and PE performed literature review, PE created the figures and PE, LP and CB contributed to the writing and revising of this manuscript.
The authors declare that they have no conflict of interest.
This research was funded by the Institut Pasteur, the Agence National de Recherche [grant number ANR-10-LABX-62-IBEID] and the DARRI/Institut Carnot/Microbe et Santé [grant number INNOV-SP10-19].
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
Escoll P, Platon L, Buchrieser C. Roles of Mitochondrial Respiratory Complexes during Infection. Immunometabolism. 2019;1:e190011. https://doi.org/10.20900/immunometab20190011

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




