Location: Home >> Detail
Immunometabolism. 2022;4(2):e220007. https://doi.org/10.20900/immunometab20220007
 ,
Nikolaos Patsoukis 1,2,*
,
Nikolaos Patsoukis 1,2,*
1 Division of Hematology-Oncology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02215, USA
2 Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02215, USA
* Correspondence: Vassiliki A. Boussiotis, Tel.: +1-617-667-8563; Nikolaos Patsoukis, Tel.: +1-617-667-8573.
Programmed Death-1 (PD-1; CD279) is an inhibitory receptor induced in several activated immune cells and, after engagement with its ligands PD-L1 and PD-L2, serves as a key mediator of peripheral tolerance. However, PD-1 signaling also has detrimental effects on T cell function by posing breaks on antitumor and antiviral immunity. PD-1 blocking immunotherapy either alone or in combination with other therapeutic modalities has shown great promise in cancer treatment. However, it is unclear why only a small fraction of patients responds to this type of therapy. For this reason, efforts to better understand the mechanisms of PD-1 function have recently been intensified, with the goal to reveal new strategies to overcome current limitations. The signaling pathways that are inhibited by PD-1 impact key regulators of metabolism. Here, we provide an overview of the current knowledge about the effects of PD-1 on metabolic reprogramming of immune cells and their consequences on systemic metabolism.
PD-1 and its ligands are components of a central inhibitory pathway upregulated upon activation in several cell types of the adaptive and innate immune system. Its main function is to prevent autoimmunity while keeping immune responses efficient and balanced [1–4]. However, PD-1 signaling prevents antitumor immunity by interacting with its ligands expressed on cancer [5] and antigen presenting cells (APC) [6,7] within the tumor microenvironment (TME). These findings led to the development of blocking therapeutic antibodies against PD-1 and its ligands that revolutionized cancer immunotherapy [8–10]. The limited success of this therapeutic approach has recently sparked extensive efforts to better understand the mechanisms of PD-1 signaling and function with the goal to improve the therapeutic success.
Besides the recent advancements in understanding the molecular mechanisms of its function (extensively reviewed in [11] and [12]), efforts have focused in understanding metabolic implications of PD-1 signaling. Extensive studies have documented that PD-1 is a central regulator of a metabolically dysfunctional state termed “exhaustion” which occurs in T cells under conditions of persistent activation as in the case of cancer and chronic viral infections [13]. Nevertheless, genetic deletion of PD-1 not only was insufficient to rescue cells from this metabolic dysfunction, but actually promoted accumulation of terminally differentiated exhausted CD8+ T cells [14]. This finding suggested that overcoming exhaustion cannot be achieved solely by targeting PD-1, since a plethora of other inhibitory receptors, such as LAG-3, CD244 (also known as 2B4), CD160, CTLA-4 and TIM-3 [15–18] as well as factors in the TME beyond checkpoint inhibitors can contribute to this dysfunctional metabolic state [19]. By altering TCR signaling, PD-1 results in alterations of immunometabolic reprogramming thereby imprinting transcriptional, metabolic and epigenetic implications on the differentiation and function of immune cells. Here, we provide a brief overview of the current knowledge regarding the effects of PD-1 on the metabolic features of immune cells.
Signaling and metabolism are two tightly linked entities. Signaling can cause metabolic changes, whereas, conversely, metabolic changes can alter the expression of signaling molecules through epigenetic and transcriptional control. PD-1 is a surface inhibitory receptor upregulated on activated immune cells and acts at the forefront of immune signaling.
T Cells PD-1 impact on T cell signaling pathways and link with metabolismPD-1 has been mostly studied in T cells and upregulation of PD-1 expression is tightly correlated with T cell activation. TCR signaling induces PD-1 expression through the transcriptional activator NFATc1 [20] (Figure 1). It has been shown that artificial increase of basal TCR signaling by expressing the semi-active mutant form of ZAP70 W131A, which disrupts normal ZAP70 autoinhibition, triggered a feedback increase in inhibitory receptor expression including PD-1 and resulted in T cell unresponsiveness, partially reversible by PD-1 blockade [21]. In addition, PD-1 upregulation in T cells can be induced by the Notch signaling pathway [22]. PD-1 ligation by its ligands PD-L1 or PD-L2, concomitantly with TCR signaling, results in Src-family kinase (Lck and/or Fyn)-mediated tyrosine phosphorylation of the PD-1 cytoplasmic tail at the immunoreceptor tyrosine inhibitory motif (ITIM) and at the immunoreceptor tyrosine switch motif (ITSM) leading to the recruitment of phosphatase SHP-2 which then downregulates signaling pathways associated with the T cell receptor (TCR) and the CD28 costimulatory receptor [23–36] (Figure 1A). Two main target pathways include Ras/MAPK and PI3K/Akt/mTOR, which are key regulators of T cell metabolism, primarily integrating signals from the TCR, costimulatory and cytokine receptors to promote a glycolytic phenotype [37,38] (Figure 1A). PD-1 counteracts these pathways, and keeps T cells “on a diet” as it decreases glycolysis, amino acid transport and metabolism [27,39]. Concomitantly, PD-1 signaling promotes fatty acid oxidation (FAO) of endogenous lipids by increasing expression of carnitine palmitoyl-transferase 1A (CPT1A), and induces lipolysis by increasing expression of adipose triglyceride lipase (ATGL) [39] (Figure 1B).
Notably, PD-1 engagement can also increase polyunsaturated fatty acids (PUFA) intracellularly and in culture supernatants [39]. PUFAs can inhibit T cell proliferation and interleukin-2 (IL-2) production in vitro and in vivo [40,41]. Due to the presence of two or more carbon–carbon double bonds PUFA are sensitive to oxidation by reactive oxygen species (ROS). Notably, uptake of oxidized lipids by the scavenger receptor CD36 can promote lipid peroxidation and dysfunction of CD8+ T cells in tumors [42]. Thus, PD-1-mediated increase of PUFA may be an important mechanism contributing to T cell dysfunction by increasing oxidized lipids in the TME.
Little is known about the effects of PD-1 on the redox metabolism of T cells. Metabolomics analysis of T cells subjected to PD-1 ligation showed decreased levels of reduced glutathione GSH and higher levels of cysteine-GSH disulfide indicative of a more oxidative environment in T cells receiving PD-1 signals [39]. A recent study found that alloreactive T cells from a mouse graft versus host disease (GVHD) model had increased expression of PD-1 and FAO-derived ROS, which made the cells more susceptible to F1F0-ATP synthase complex inhibitors [43]. These effects were reverted either by antioxidants or by PD-1 blockade, resulting in decreased cellular ROS, making the cells resistant to F1F0-ATP inhibition [43]. In contrast, other investigators reported that PD-1 blockade increased cellular ROS and mitochondrial mass together with proliferation and activation of CD8+ T cells in the tumor microenvironment [44].
Another pathway involved in T cell metabolism that is targeted by PD-1, particularly in regulatory T cells (Treg), is the TGF-β pathway. TGF-β counteracts the PI3K pathway and promotes FAO to fuel mitochondrial oxidative phosphorylation (OXPHOS) [45]. PD-1 favors this metabolic phenotype [39]. In parallel, PD-1 reduces the threshold of TGF-β-mediated Treg development [46] while enhancing SMAD3 transactivation [31] (Figure 1B). TGF-β attenuates anti-tumor responses mediated by PD-L1 blocking immunotherapy in patients and murine tumor models by promoting a T cell exhaustion phenotype. Conversely, simultaneous blockade of PD-1 and TGF-β-mediated signaling, improved responses in patients with various cancers [47]. Notably, recent preclinical studies employing bifunctional fusion proteins blocking the PD-1 pathway in conjunction with TGF-β showed significant enhancement of antitumor activity [48,49] and such outcomes were also confirmed in patients [50,51].
Another mechanism promoting PD-1-mediated T cell dysfunction in chronic infections, such as HIV in humans and LCMV in mice, is the upregulation of the basic leucine zipper ATF-like transcription factor (BATF) [52]. Interestingly, BATF was found to couple triglyceride (TG) metabolism with Treg suppressive function for controlling allergic airway inflammation and IgE responses [53]. However, the role of BATF in T cell exhaustion remains controversial, as recent studies supported a beneficial role of BATF on T cell responses in chronic infection [54] and cancer [55].
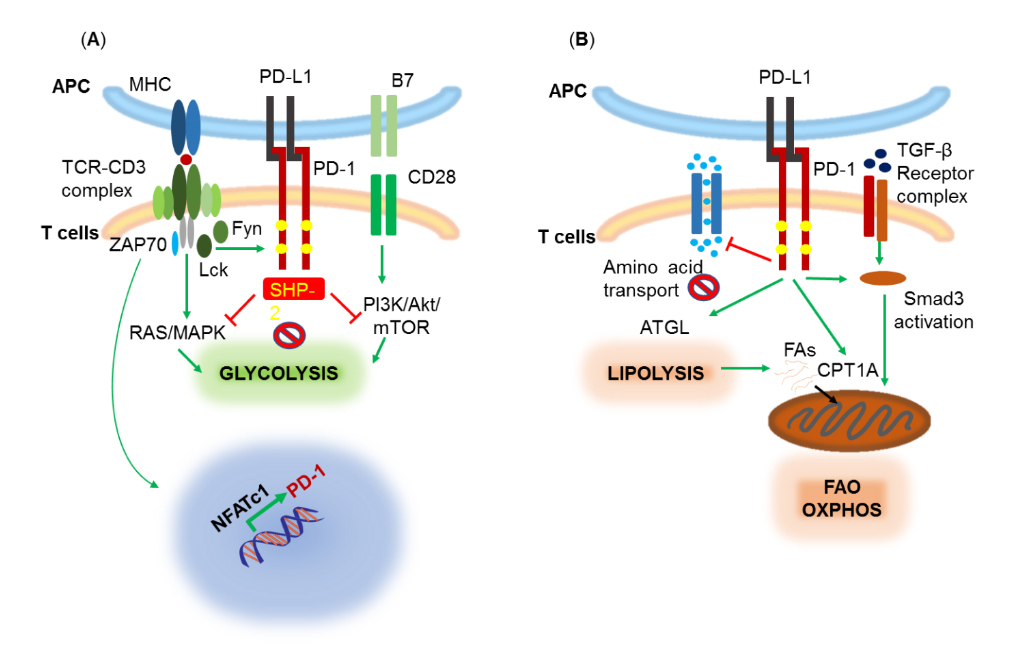 Figure 1. Effect of PD-1 on T cell-mediated signaling pathways and metabolism. (A) TCR stimulation induces NFATc1-mediated PD-1 upregulation. PD-1 is phosphorylated by TCR-proximal Src-family kinases Lck and Fyn and recruits SHP-2 phosphatase which downregulates TCR- and CD28-mediated signals thereby preventing induction of glycolysis by these pathways. (B) Besides, glycolysis, PD-1 inhibits amino acid transport and metabolism and promotes lipolysis and FAO by inducing expression of ATGL and CPT1A, respectively. PD-1 also enhances the TGF-β signaling through Smad3 activation, a pathway known to promote FAO, OXPHOS, and Treg differentiation.
Figure 1. Effect of PD-1 on T cell-mediated signaling pathways and metabolism. (A) TCR stimulation induces NFATc1-mediated PD-1 upregulation. PD-1 is phosphorylated by TCR-proximal Src-family kinases Lck and Fyn and recruits SHP-2 phosphatase which downregulates TCR- and CD28-mediated signals thereby preventing induction of glycolysis by these pathways. (B) Besides, glycolysis, PD-1 inhibits amino acid transport and metabolism and promotes lipolysis and FAO by inducing expression of ATGL and CPT1A, respectively. PD-1 also enhances the TGF-β signaling through Smad3 activation, a pathway known to promote FAO, OXPHOS, and Treg differentiation.
Besides suppressing activation and IL-2 production by T effector cells, thereby inducing a state of anergy [56], PD-1 also affects T cell memory differentiation as T cells from PD-1 knockout mice are skewed towards an effector (TEM) over a central (TCM) memory phenotype [57]. By using single-cell RNA-sequencing, a recent study identified two previously unrecognized distinct stem-like CD8+ memory T cell subsets with distinct fate commitments in humans, one lacking PD-1 and T cell immunoreceptor with Ig and ITIM domains (TIGIT) committed to a functional lineage, and a second expressing PD-1 and TIGIT committed to a dysfunctional, exhausted-like lineage [58]. However, it was previously observed that complete lack of PD-1 signaling was not beneficial for long-term memory differentiation, but resulted in the generation of exhausted, short lived, terminally differentiated CD8+ T cells [14]. Notably, recent reports documented that PD-1 plays a critical role in efficient memory differentiation by preserving a key memory CD8+ T cell precursor pool during initial activation, and supporting its persistence after antigen clearance [59,60]. Collectively, PD-1 acts as a key metabolic regulator of T cell memory, a critical function that should be taken into consideration when PD-1 blockade therapy is designed.
The role of PD-1 on Treg function and metabolismAs mentioned above, PD-1 promotes Treg differentiation by lowering the threshold of TGF-β signaling [46]. Upregulation of PD-1 and other inhibitory receptors on tumor-infiltrating Treg cells had been correlated with their enhanced suppressive function [61]. A surprising recent finding was that PD-1 blockade, besides inducing recovery of dysfunctional PD-1+ T cells, enhances the immunosuppressive function of PD-1+ Treg thereby promoting cancer hyperprogression [62,63]. These observations suggest a negative impact of PD-1 on Treg suppression activity and indicate that the balance of PD-1 expression between T effector and Treg cells may be a predictive biomarker of the clinical efficacy of PD-1 blocking immunotherapy [63]. Notably, in the tumor microenvironment, Treg cells but not FoxP3−CD4+ or CD8+ T cells have the ability to utilize lactic acid produced by highly glycolytic cancer cells, through MCT1-mediated import, conferring metabolic advantage and survival. Lactic acid-driven metabolism and activation of Treg in the tumor microenvironment results in preferential upregulation of PD-1 in Treg cells leading to imbalanced PD-1 expression between T effector and Treg and impairment of PD-1 blocking immunotherapy [64].
Specific effects of PD-1 on Treg function and metabolism had not been directly assessed until recently, when mice with Treg-specific PD-1 deletion were generated [65]. In agreement with previous observations [62,63] the study found that lack of PD-1 expression in Treg cells increased their suppressive function and demonstrated the in vivo significance of this effect by the improved outcomes in mouse models of autoimmunity, such as experimental autoimmune encephalomyelitis (EAE) and non-obese diabetes (NOD) selectively lacking PD-1 in Treg cells [65]. Since PD-1 engagement inhibits the PI3K/Akt pathway on effector T cells [26,31,46], and promotes lipid oxidation [39], a preferred metabolic pathway in Treg cells [66], it would be anticipated that genetic ablation of PD-1 in Treg would enhance activation of the PI3K/Akt pathway and glycolysis. In contrast, PD-1-deficient Treg cells were found to have decreased glycolytic activity due to reduced PI3K/Akt/mTOR signaling [65]. This finding, although surprising, could explain the enhanced suppressive activity of PD-1-deficient Treg as inhibition of the PI3K/Akt/mTOR pathway is necessary for the suppressive function of Treg cells [67,68]. In addition, activated PD-1-deficient Treg cells had increased mitochondrial spare respiratory and maximal respiratory capacity and displayed a bioenergetic state characterized by enhanced mitochondrial function but reduced glycolysis [65]. The mechanisms of these effects of PD-1 ablation in Treg remain elusive. Collectively, the role of PD-1 is to inhibit the function of T effector cells and Treg cells, and genetic ablation or blockade of PD-1 can reverse this outcome, albeit by inducing opposite metabolic signatures in these two cell types (Figure 2).
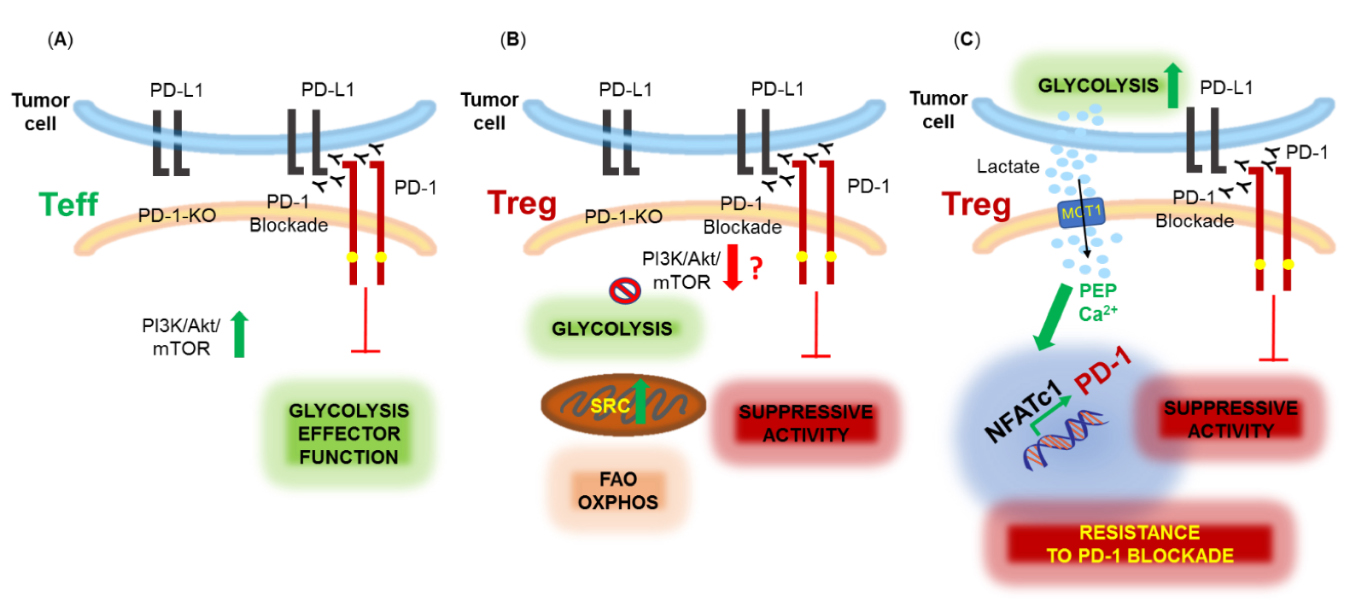 Figure 2. Metabolic effects of PD-1 ablation or blockade in effector (Teff) and regulatory (Treg) T cells. (A) PD-1 inhibits the effector function of Teff cells which is restored by PD-1 genetic ablation or blockade, promoting activation of the PI3K/Akt/mTOR pathway and glycolysis. (B) PD-1 blockade or ablation enhances the suppressive function of Treg, possibly, by downregulating the PI3K/Akt/mTOR pathway and glycolysis and promoting mitochondrial activity and spare respiratory capacity (SRC). (C) Highly glycolytic tumors produce high levels of lactic acid (lactate) in the TME. Lactate is uptaken mainly by Treg cells expressing monocarboxylate transporter 1 (MCT1) and converted into phosphoenol pyruvate (PEP) leading to increased calcium (Ca2+) in the cytoplasm, NFATc1 translocation into the nucleus and upregulation of PD-1 expression. PD-1 blockade under conditions leading to this metabolic adaptation in Treg cells enhances Treg suppressive function resulting in resistance to PD-1 blockade therapy.
Figure 2. Metabolic effects of PD-1 ablation or blockade in effector (Teff) and regulatory (Treg) T cells. (A) PD-1 inhibits the effector function of Teff cells which is restored by PD-1 genetic ablation or blockade, promoting activation of the PI3K/Akt/mTOR pathway and glycolysis. (B) PD-1 blockade or ablation enhances the suppressive function of Treg, possibly, by downregulating the PI3K/Akt/mTOR pathway and glycolysis and promoting mitochondrial activity and spare respiratory capacity (SRC). (C) Highly glycolytic tumors produce high levels of lactic acid (lactate) in the TME. Lactate is uptaken mainly by Treg cells expressing monocarboxylate transporter 1 (MCT1) and converted into phosphoenol pyruvate (PEP) leading to increased calcium (Ca2+) in the cytoplasm, NFATc1 translocation into the nucleus and upregulation of PD-1 expression. PD-1 blockade under conditions leading to this metabolic adaptation in Treg cells enhances Treg suppressive function resulting in resistance to PD-1 blockade therapy.
Although PD-1 has been mostly studied in T cells, it is increasingly being appreciated that is also upregulated in other immune cell types including innate immune cells such as macrophages [69–71], tumor-associated macrophages (TAMs) [72], monocytes [73,74] myeloid progenitors and myeloid-derived suppressor cells (MDSC) [75] and innate lymphoid cells (ILCs) such as natural killer (NK) cells [76], and regulates their differentiation and function as previously extensively reviewed [11,77-79].
Myeloid cellsLimited information is currently available regarding the effects of PD-1 on the signaling and metabolic reprogramming of myeloid cells. A recent study examining immune dysfunctions in chronic lymphocytic leukemia (CLL) found that PD-1 triggering on monocytes resulted in impaired glycolysis, phagocytosis and Bruton's tyrosine kinase (BTK) signaling and that blocking the PD-1 pathway reverted the metabolic dysfunction [74] (Figure 3A).
It was recently shown that granulocyte/macrophage progenitors (GMPs), which accumulate during cancer-driven emergency myelopoiesis and give rise to myeloid-derived suppressor cells (MDSCs), express PD-1 and PD-L1 [75]. Myeloid-specific ablation of PD-1 in tumor-bearing mice, prevented MDSC accumulation, while increasing systemic output of effector myeloid cells and T effector memory cells with improved antitumor function. These outcomes were correlated with metabolic alterations in PD-1-deficient myeloid progenitors in response to factors that drive emergency myelopoiesis characterized by increased intermediates of glycolysis, pentose phosphate pathway, and TCA cycle but, most prominently, elevated cholesterol, a key mediator of inflammatory differentiation of macrophages and DC favoring antigen-presenting function [80] (Figure 3B). Thus, skewing myeloid differentiation towards effector and inflammatory antigen presenting cells rather than immunosuppressive MDSC might be a key mechanism of antitumor immunity mediated by PD-1 blockade.
NK cellsNK cells form an important component of the innate lymphoid system [81,82]. Despite their lack of antigen-specific receptors they have significant antiviral and antitumor activity through antigen-non-specific mechanisms and, similarly to T cells, their effector function relies on activation of the PI3K/Akt/mTOR pathway and glycolysis [83,84]. Upon activation, NK cells, and other ILCs, upregulate PD-1 [76]. However little is known about the role of PD-1 on the metabolic properties on NK cells. Similarly to T cells, PD-1 has an inhibitory role in NK cell function. In the context of several human cancers and mouse tumor models, PD-1 expression in NK cells has been correlated with a dysfunctional, exhausted phenotype, amenable to reversion by PD-1 blockade [85–88]. However, recent studies provided evidence for a different role of NK-expressed PD-1 in controlling host resistance to viral infection. Endogenous glucocorticoids are essential steroid hormones produced after activation of the hypothalamic-pituitary-adrenal (HPA) neuroendocrine axis in response to infection. They bind to cytosolic glucocorticoid receptors (GR) thereby mediating signals that lead to resolution of inflammation and maintenance of immune homeostasis. A recent study in a mouse model provided evidence that GR-mediated signaling in response to CMV infection induced tissue-specific PD-1 expression on NK cells. GR-mediated PD-1 expression diminished the production of IFN-γ by in NK cells and prevented lethal immunopathology without compromising viral clearance [89]. A study in human tumor samples showed that glucocorticoids present in the TME in combination with inflammatory cytokines IL-12, IL-15, and IL-18 increased PD-1 expression on NK cells and induced an immunosuppressive milieu [90]. These studies revealed a new mechanism by which PD-1 expression in innate immune cells is controlled by a neuroendocrine-immune axis, thereby regulating immune homeostasis but also compromising anti-tumor immunity.
These evolving observations reveal that PD-1 has a previously unappreciated role in the innate immune system by affecting several properties and functions of innate immune cells including metabolic and differentiation programs.
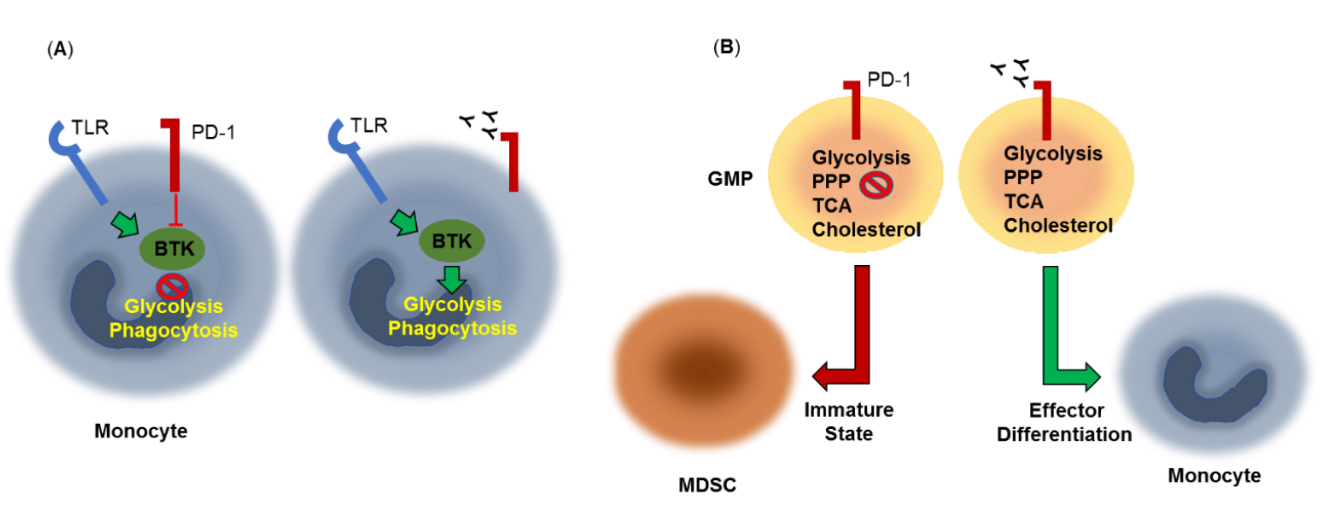 Figure 3. Metabolic effects of PD-1 in myeloid cells. (A) PD-1 triggering on monocytes results in impaired Bruton's tyrosine kinase (BTK) signaling, glycolysis and phagocytosis. PD-1 pathway blockade reverts this metabolic dysfunction. TLR: Toll-Like Receptor. (B) Granulocyte/macrophage progenitors (GMPs), which accumulate during cancer-driven emergency myelopoiesis express PD-1, which compromised growth factor-mediated signaling and metabolic activity, preserving an immature state and giving rise to myeloid-derived suppressor cells (MDSCs). PD-1 ablation or blockade prevents MDSC accumulation, and promotes effector differentiation. These effects are correlated with increased metabolic activity of glycolysis, pentose phosphate pathway (PPP), and TCA cycle and elevated cholesterol, a key enhancer of antigen-presenting function.
Figure 3. Metabolic effects of PD-1 in myeloid cells. (A) PD-1 triggering on monocytes results in impaired Bruton's tyrosine kinase (BTK) signaling, glycolysis and phagocytosis. PD-1 pathway blockade reverts this metabolic dysfunction. TLR: Toll-Like Receptor. (B) Granulocyte/macrophage progenitors (GMPs), which accumulate during cancer-driven emergency myelopoiesis express PD-1, which compromised growth factor-mediated signaling and metabolic activity, preserving an immature state and giving rise to myeloid-derived suppressor cells (MDSCs). PD-1 ablation or blockade prevents MDSC accumulation, and promotes effector differentiation. These effects are correlated with increased metabolic activity of glycolysis, pentose phosphate pathway (PPP), and TCA cycle and elevated cholesterol, a key enhancer of antigen-presenting function.
Releasing the PD-1 break on immune cells not only affects the targeted cells but can also have broader systemic impact on the metabolic regulation of the whole organism. For example, one of the first observations regarding the role of PD-1 in preventing autoimmunity was that PD-1 loss or blockade promoted the development of type 1 diabetes in mice [91,92]. Other early studies showed that impairment of the PD-1 pathway enhanced and exacerbated the development of atherosclerotic lesion in proatherogenic mice [93,94]. Recently, similar effects were observed in patients treated with PD-1 blocking checkpoint immunotherapy [95].
The systemic consequences of the metabolic alterations related to ablation of PD-1 signaling were extensively examined by analyzing water-soluble metabolites in the serum of PD-1 deficient mice, which develop a spontaneous activated phenotype [96]. Differences were observed in several metabolites related to energy-generating pathways, such as the TCA cycle, glucose and amino-acid metabolism. The most significant change affected the abundance of the aromatic amino acids tryptophan, tyrosine and phenylalanine, which were much lower in the PD-1 deficient mice compared to control, by 2 months of age. This was due to the increased intracellular transport and utilization of these amino acids by activated T cells and resulted in depletion of tyrosine and tryptophan from the brain, where these are essential for synthesis of the neurotransmitters dopamine (DA) and serotonin (5-hydroxytryptamine (5-HT)). The consequence was a substantial deficiency in both these neurotransmitters in the brain, resulting in behavioral changes associated with anxiety behavior and exacerbated fear responses [96].
Systemic metabolic adaptations to PD-1 blockade immunotherapy were also detected in human patients. Comprehensive analysis of serum metabolites in patients with advanced melanoma and renal cell carcinoma treated with the anti-PD-1 antibody nivolumab, identified increase of kynurenine/tryptophan ratio. This finding, indicative of increased activity of indoleamine 2,3-dioxygenase (IDO), was associated with worse overall survival, suggesting that increase of IDO activity after PD-1 checkpoint immunotherapy might be a mechanism of systemic adaptive immune resistance [97]. Conversely, as determined by a different study using samples from patients treated with checkpoint immunotherapy, elevated metabolites derived from the microbiome (hippuric acid), fatty acid oxidation (butyrylcarnitine) and redox metabolism (cystine and glutathione disulfide), provided high response probability. In the same patients, systemic increase of CD4+ and CD8+ T cells combined with elevated expression of PGC-1 and ROS were also correlated with improved responses to nivolumab [98].
Collectively, these findings suggest that interfering with PD-1 function on immune cells may have consequences that extend beyond the immune system. Moreover, metabolic monitoring might have a predictive value of therapeutic response and might assist in patient stratification for combining metabolic interventions with PD-1 blockade immunotherapy. However, much remains to be learned regarding the effects of PD-1 signaling or its blockade on systemic metabolic adaptation in the context of cancer. Based on the very limited available reports in the literature [96–98] PD-1 blockade resulted in distinct metabolic alterations in the context of different tumors. For example, although activated T cells appeared to have a dominant role in altering systemic metabolic responses in the context of a murine tumor model [96], in human patients treated with checkpoint immunotherapy, metabolic alterations of cancer cells dominated the systemic metabolic outcome [97]. Additional work in human patients and mouse tumor models is required to dissect the determinants of systemic metabolic adaptations to checkpoint immunotherapy and guide rational approaches of systemic metabolic interventions to enhance the therapeutic outcome of checkpoint immunotherapy.
It is increasingly being appreciated that besides T cells, PD-1 engagement or blockade have a major impact on metabolic reprogramming of multiple immune cell types and these effects can extend systemically. Although signaling- and metabolism-targeting effects of PD-1 engagement and blockade have been extensively studied in T cells, evolving studies provide evidence for the important implications of PD-1 signaling in several cell types of the innate immune system. Besides metabolomics and bioenergetics on sorted cell populations, novel methodologies such as SCENITH [99] will allow a more accurate metabolic characterization and understanding of PD-1 effects on rare immune populations. It is now apparent that targeting the PD-1 pathway alone is not sufficient to overcome the complex dysfunctional state of various immune cell populations in the tumor microenvironment. Harnessing metabolism combined with interventions to checkpoint pathways holds a great promise against this challenge.
VAB has patents on the PD-1 pathway licensed by Bristol-Myers Squibb, Roche, Merck, EMD-Serono, Boehringer Ingelheim, AstraZeneca, Novartis, and Dako. The authors declare no other competing interests.
This research was funded by NIH grants R01CA212605, R01CA229784, and R01CA238263 (to VAB); R21AR073494 (to NP).
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
Boussiotis VA, Patsoukis N. Effects of PD-1 Signaling on Immunometabolic Reprogramming. Immunometabolism. 2022;4(2):e220007. https://doi.org/10.20900/immunometab20220007

Copyright © 2022 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




