Location: Home >> Detail
Immunometabolism. 2021;3(3):e210021. https://doi.org/10.20900/immunometab20210021

1 University of Ottawa Heart Institute, 40 Ruskin St, Ottawa, ON, K1Y 4W7, Canada
2 Department of Biochemistry, Microbiology and Immunology, Faculty of Medicine, University of Ottawa, 451 Smyth Road, Ottawa, ON, K1H 8M5, Canada
* Correspondence: Mireille Ouimet.
This article belongs to the Virtual Special Issue "Cardiovascular Immunometabolism"
A hallmark of sterile and nonsterile inflammation is the increased accumulation of cytoplasmic lipid droplets (LDs) in non-adipose cells. LDs are ubiquitous organelles specialized in neutral lipid storage and hydrolysis. Originating in the ER, LDs are comprised of a core of neutral lipids (cholesterol esters, triglycerides) surrounded by a phospholipid monolayer and several LD-associated proteins. The perilipin (PLIN1-5) family are the most abundant structural proteins present on the surface of LDs. While PLIN1 is primarily expressed in adipocytes, PLIN2 and PLIN3 are ubiquitously expressed. LDs also acquire a host of enzymes and proteins that regulate LD metabolism. Amongst these are neutral lipases and selective lipophagy factors that promote hydrolysis of LD-associated neutral lipid. In addition, LDs physically associate with other organelles such as mitochondria through inter-organelle membrane contact sites that facilitate lipid transport. Beyond serving as a source of energy storage, LDs participate in inflammatory and infectious diseases, regulating both innate and adaptive host immune responses. Here, we review recent studies on the role of LDs in the regulation of immunometabolism.
Lipid metabolism is defined as the synthesis and degradation of lipids, including the breakdown or storage of fats for energy homeostasis and the synthesis of structural and functional lipids. Lipid droplets (LDs), dynamic and ubiquitous organelles that regulate the storage and hydrolysis of lipids, have emerged as key mediators of lipid metabolism, with abnormal LD dynamics having been linked to the pathophysiology of metabolic and inflammatory diseases, neurodegeneration and aging. With LDs at the center stage of lipid metabolic disorders and biofuel production, LD research has become an exciting field of cell biology. Here, we discuss how the dynamic cycle of LD biogenesis and catabolism regulates the availability of lipids that sustain cell metabolism or serve as precursors of signalling mediators, highlighting a critical role for LDs in integrating metabolic and inflammatory processes in immune cells.
Morphologically, LDs are composed of a neutral core rich in triglycerides (TG) and cholesterol-esters (CE) enclosed by a phospholipid monolayer and surrounded with proteins serving structural, metabolic, regulatory, and membrane-trafficking functions. Originating at the ER, the mechanisms underlying LD biogenesis remain under investigation. The prevailing model suggests that this occurs in 3 stages: neutral lipid synthesis, lens formation, and budding [1,2]. LD biogenesis is initiated by the synthesis and accumulation of neutral lipids between the leaflets of the ER bilayer, where they remain dispersed at low concentration. Their eventual buildup leads to the phase separation of neutral lipids into a lens structure, believed to be guided by the principles of physical chemistry that favor energy conservation by limiting lipid interactions [3,4]. Small and short-lived, the lens structures have curiously remained theoretical for a long time, until Choudhary et al. visualized the phenomenon using electron tomography in yeast in 2015, thus finally providing evidence to support the elusive mechanism of LD emergence from the ER outer leaflet into the cytoplasm [5].
In parallel to neutral lipid synthesis, phospholipid synthesis is induced at the ER during LD biogenesis to provide the building blocks of the LD surface monolayer. With phosphatidylcholine (PC) as the primary component of this monolayer, LD biogenesis requires de novo PC synthesis by the cytidine diphosphate (CDP)-choline pathway [6]. This is regulated by translocation of CTP:PC cytidylyltransferases CCTα and CCTβ to growing LDs [6,7]. The recruitment of seipin oligomers and LDAF1, and the coating of LDs with perilipin proteins facilitates LD growth and budding [8–12]. LDs also acquire a host of enzymes and proteins that regulate LD metabolism [13]. The dynamic and interactive nature of LDs along with the variability in metabolic demands of distinct cell types results in a high diversity in both the structural organization and lipid and protein content of LDs [14]. For example, LDs from adipocytes contain TG-enriched cores, whereas CE enrichment is observed in foamy macrophages [15].
Adding to this diversity, studies investigating the proteome of LDs have emphasized their complex and varied protein composition. The perilipin (PLIN) family are the most abundant structural proteins present on the surface of LDs, with regulatory roles in lipolysis for the mobilization of fats [16]. Five members of this family have been identified, with differential expression in mammalian tissues. PLIN1 is primarily expressed in adipocytes, while PLIN2 and PLIN3 are ubiquitously expressed and finally PLIN4 and PLIN5 expression is restricted to other tissues. Importantly, PLIN1 phosphorylation is required to mobilize lipids in adipose tissue for lipolysis by neutral lipases adipose triglyceride lipase (ATGL), hormone sensitive lipase (HSL) and monoacylglycerol (MAGL) [17]. The exact mechanism through which each PLIN member regulates lipolysis remains to be fully elucidated. Nevertheless, it is now evident that in addition to lipid homeostasis, diverse cellular functions can be influenced by LD surface proteins. Trafficking proteins from the SNAP REceptor (SNARE) and member RAS oncogene family (RAB) families are also present on LDs, indicative of the propensity of LDs for fusion events as well as transient interactions. LD interactions with the ER, endosomes, mitochondria and peroxisomes are well documented, involving the PLIN proteins in many instances as mediators of these interactions [18]. These dynamic LD contacts enable both lipid and protein exchanges, and contribute to the regulation of lipid metabolism, energy homeostasis, membrane trafficking and nuclear function. While the cycle of biogenesis and catabolism of LDs can be observed in most cells, imbalances in the turnover of these organelles and their consequential accumulation is linked to many pathological conditions, including sterile and nonsterile inflammation.
Cytoplasmic LD turnover is a highly regulated process. Best characterized is the hydrolysis of neutral lipids in the LD core through the action of cytoplasmic lipases, called lipolysis. Through the sequential action of cytosolic lipases ATGL, HSL and MAGL that act at a neutral pH, free fatty acids (FFAs), glycerol, and intermediates (mono- and diglycerides) are released from TG stored in LDs [19]. In turn, cytoplasmic CE hydrolases such as neutral cholesterol ester hydrolase 1 (NCEH1), carboxylesterase 1 (CES1) and HSL promote LD CE hydrolysis and the release of free cholesterol from the LD core [15]. The enzymatic activity of lipases at the LD surface is regulated by signalling events and protein interactions notably involving PLIN1, PLIN2 and PLIN5 [17].
In addition to lipolysis, autophagy-mediated LD catabolism significantly contributes to the degradation of LDs. Autophagy-dependant LD breakdown is best described in hepatocytes [20] and macrophage foam cells [21,22], while its role in other tissues such as adipose tissue is less understood [23,24]. From the Ancient Greek auto (self) and phagein (to eat), autophagy is the natural mechanism through which cells digest damaged organelles and cytoplasmic components for later recycling. There are 3 autophagy subtypes: microautophagy, macroautophagy and chaperone-mediated autophagy (CMA). Selective autophagic degradation of LDs, distinct from bulk autophagy, has been coined lipophagy [20]. Evidence points to direct roles for both macroautophagy and microautophagy in lipophagy [20,22,25], while CMA can facilitate LD breakdown through the removal of PLIN proteins [20–22,26]. In microlipophagy, lysosomal membranes directly enwrap and transport LDs into the lysosome lumen, a process that is dependent contact sites between LDs and lysosomes to facilitate inter-organelle lipid transfer [25]. In macrolipophagy, double-membrane autophagosomes engulf entire LDs or fractions of LDs [20], and subsequent fusion of LD-containing autophagosomes with lysosomes results in degradation of LD proteins and lipids by lysosomal proteases and lysosomal acid lipase (LAL) respectively.
More recently, clusters of LDs were also shown to be tagged for selective autophagic degradation by an aggrephagy-like mechanism [22,27], and indeed several mediators of aggrephagy, including optineurin (OPTN), next to BRCA1 gene 1 protein (NBR1), heat shock protein family A member 5 (HSPA5), valosin containing protein (VCP/TERA) and YWHA/14-3-3 proteins localize to LDs [22]. Further research is needed to elucidate the cellular mechanisms of lipophagy and determine the specific contributions of macroautophagy, microautophagy and CMA to lipophagy of single or clusters of LDs. Growing evidence supports the notion that lipolysis and lipophagy are not mutually exclusive and can occur in tandem [28]. In fact, a size-dependant regulation of LD fate was recently described in hepatocytes, where larger LDs were preferentially targeted by cytosolic neutral lipases whereas smaller lipid droplets underwent lipophagy [29]. Together, neutral lipolysis and lipophagy release FFAs and cholesterol molecules from LDs. FFAs are mainly shuttled to mitochondria to fuel energy metabolism or can act in a signalling capacity. Genomic regulators such as the transcription factor EB (TFEB) related to LD catabolism also regulate the expression of genes involved in autophagy, lysosome biogenesis and mitochondrial FFA β-oxidation, coupling LD turnover with associated bioenergetic programs and inflammation [30,31]. On the other hand, LD-derived free cholesterol can be (i) exported via ATP-binding cassette cholesterol (ABC) transporters or, in what is referred to as the “futile LD cholesterol cycle” or (ii) re-esterified at the ER for repackaging and storage in LDs [15].
First reported in 2013, nuclear lipid droplets (nLDs) are less frequent and smaller than their cytosolic counterparts [32–35]. nLDs contribute to ~2–10% of lipids stored in LDs in hepatocytes treated with oleic acid [35]. Though their origin and physiological significance remains poorly defined, nLDs appear to primarily associate with the type I nucleoplasmic reticulum (type I NR) and promyelocytic leukemia nuclear bodies (PML NBs) [36]. The type I NR results from an invagination of the inner nucleoplasmic membrane, while PML NBs are membrane-less macromolecular multiprotein complexes within the nucleoplasm formed by PML proteins. In cells where the PML isoform PML-II localises to the type I NR, nLDs are frequent, at a density that directly correlates to the level of PML-II expression [36–38]. nLDs share compositional similarities with cytosolic LDs (e.g., presence of PLIN3, Rab18), but significant disparities have also been observed (e.g., absence of PLIN2), warranting further proteomic and lipidomic characterisation of these organelles [32,36].
Sołtysik et al. linked the origin of nLDs to the mechanisms responsible for very-low density lipoprotein (VLDL) synthesis [38]. In hepatocytes, the directional budding of nascent LDs towards the cytoplasm yields cytosolic LDs, while budding into the ER lumen gives rise to luminal LDs, the precursors of VLDL particles. Formation of luminal LDs is driven by the microsomal triglyceride transfer protein (MTP) that contributes to secretion-competent ApoB-containing VLDL particles as well as ApoB-deficient luminal LDs [39–41]. Under conditions where ApoB expression is decreased (e.g., ER stress) [42,43] or absent (e.g., enterocytes) [44], MTP drives the production of ApoB-free luminal LDs that accumulate in the ER lumen [41]. Sołtysik et al. propose that this accumulation generates large LDs in the type I NR that relocate to the nucleoplasm [38] and that nLDs mitigate ER stress by recruiting CCTα to enhance PC synthesis and expansion of the ER. This process may be regulated by the competitive binding of PLIN3 and CCTα to nucleoplasmic LDs [38]. Moreover, Sołtysik et al. proposed that the requirement for the formation of a type I NR and for the lipoprotein synthesis machinery may justify why nLDs are readily observed in hepatocytes in contrast to other tissues. Interestingly, both these requirements have been documented in some immune subsets, including macrophages [45–47].
Subsequent studies have investigated nLDs in non-hepatocytes [37,48]. In an osteosarcoma cell line, the inner nuclear membrane was found to possess metabolic activity enabling nLDs genesis in situ by harbouring the enzymes required for TG synthesis [48,49]. Formation of nLDs was inversely correlated with the expression of seipin, an important contributor to cytosolic LD formation, and seipin depletion increased lipin-1β expression and nuclear phosphatidic acid synthesis [48]. While lipin-1β expression is restricted to the cytoplasm in some cells, such as adipocytes and hepatocytes, its distribution in the nucleus has been observed in other cell types such as macrophages [50]. These studies found that by mediating conversion of phosphatidic acid to diacylglycerol, lipin-1β translocation to the nucleus facilitates nLD formation. The maturation of nLDs was assisted by PML NBs association that contributes to recruit CCTα and lipin-1β [37,48].
In response to pathogenic insults including bacterial, viral, and parasitic infections, as well as endogenous stressors such as trauma or inflammation, the formation of LDs is increased in several tissues [51–57]. The frequency with which this phenomenon occurs in activated immune cells has marked LDs as a structural indicator of inflammation [58–61]. Interest in these inflammatory landmarks was amplified in recent years with the delineation of their role in both productive immune responses and immunosuppression, as well as the identification of mechanisms through which pathogens leverage LD metabolism to promote their survival [62–66]. While generally absent in resting immune cells, biogenesis of LD occurs rapidly following insult [67]. Cellular coordination and communication regulates LD numbers through lipid mediators, cytokine, and chemokine signalling, highlighting the participation of LDs in mounting the innate and adaptive immune responses [59].
In 1989, Charles Janeway Jr. proposed the idea that cells rapidly respond to constant challenges from exogenous and endogenous insults using pattern-recognition receptors (PRRs) [68,69]. Numerous PRRs grouped in five families have since been identified, functioning in cells to readily recognise and respond to pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) [70]. A role for PRRs, and predominantly the Toll-like Receptor (TLR) family, has been identified in promoting LD biogenesis during pathogenic challenge [71–75]. TLRs orchestrate innate immune responses by signalling through NF-кB, IRF and MAPK to induce interferon and pro-inflammatory cytokine production [76]. As summarized by Pereira-Dutra [61], TLR signalling induces the upregulation of enzymes involved in lipid uptake, synthesis and transport, including FASN, DGAT1/2, ACAT1/2 [73,77], and LD structural proteins, PLIN2 [72,73,78] and PLIN3 [10,79]. Furthermore, the release of cytokines following PRR stimulation results in autocrine, paracrine, and endocrine signalling events that coordinate the immune response and can induce LD biogenesis. MCP-1 [80,81], PAF [82], eotaxins [83], eicosanoids [84], TGF-β [85], RANTES [83], IL-6 [86] and interferon signalling [67,87] have all been linked to the upregulation of lipid synthesis enzymes and LD-associated proteins [59]. Because ER stress and the unfolded protein response are common consequences of pathogenic infection and inflammation, LDs may furthermore arise as a coping response to ER stress [51,56,88]. LD biogenesis contributes to removal of misfolded proteins, lipid homeostasis and management of proteins regulating cellular stress responses [51].
More recently, Hsieh et al. documented that TLR activation results in the regulated remodeling of the macrophage lipidome in a manner that is dependent on the lipid composition and metabolic state of the cell at time of stimulation [89]. These effects were oppositely driven by MyD88 and TRIF signalling pathways. As such, TLR signalling through MyD88 increased saturated and monounsaturated fatty acid synthesis in a NRF2- and SERBP1-dependent manner. TLR signalling through TRIF and autocrine IFN signalling mediated the opposite effect. Importantly, Hsieh et al. determined that different TLRs induce lipidome remodeling in distinct ways, hinting at the importance of this modulation to initiate specific pro-inflammatory programs. How these changes regulate the formation, composition, and function of LDs, and how this might modulate immune responses remains unknown. Nevertheless, this highlights the profound, stimuli-dependent lipidome remodeling initiated by PRR signalling.
The transition of immune cells from resting to activated phenotypes requires re-attribution of nutrients into the different metabolic pathways to direct or support functional changes [90–99]. With their high-energy content, LDs organize and support host immunity. LD-derived lipids can fuel critical metabolic processes or be utilized for synthesis of membranes, inflammatory mediators or signalling to the nucleus [61,100]. The general metabolic trend followed by activated immune cells points to a higher reliance on aerobic glycolysis in cells mediating inflammatory processes [99]. In opposition, cells with immune modulatory or reparative roles tend to utilize fatty acid oxidation for energy production [99]. These metabolic adaptations in response to activating signalling is nicely exemplified by the extreme polarization phenotypes of macrophages, namely pro-inflammatory M1 and pro-resolving M2 (Figure 1A) [94,95,101,102]. Upon exposure to M1 polarizing factors (e.g., TLR ligands, TNF-α, IFNγ), signalling through TLR/NF-кB [103,104] and/or AKT/mTOR [105], macrophages engage aerobic glycolysis and the pentose phosphate pathway to meet their energy requirement. These metabolic adaptations are mediated by the transcriptional regulator HIF1α, which upregulates the expression of enzymes involve in glucose uptake (e.g., GLUT1), glycolysis, conversion of pyruvate into lactate, and inflammatory mediators [106].
Remodelling of energy metabolism directly affects immune functions in a myriad of elegant cross-regulatory mechanisms [95,101]. Importantly, M1-polarized macrophages are characterised by an increase in the synthesis of fatty acids and cholesterol, regulated through the NF-кB/SREBP-1a axis [107,108]. Lipogenesis in M1 macrophages is essential to support their inflammatory and phagocytic roles, which require extensive remodelling of the plasma membrane, ER and Golgi apparatus and access to precursors to produce bioactive lipids [71]. This is substantiated by the observed decrease in phagocytosis, cytokine production and inflammasome activation in SREBP-1a-depleted macrophages [109]. In coordination with its consequential rise in intracellular fatty acids, TLR engagement facilitates the formation of LDs through upregulation of LD-associated proteins and induction of ER stress. Assembly of LDs during immune activation may therefore function dually as a buffering mechanism to control the rapid rise in lipid synthesis, providing protection against lipotoxicity, and to fuel immune responses by synthesizing and storing bioactive molecules [110]. Interestingly, the tendency for increased LD biogenesis and accumulation coincides with the pro-inflammatory roles of macrophages and the identified roles of LDs in mounting these defenses.
In contrast to M1, M2 macrophages that rely on fatty acid oxidation and OXPHOS do not accumulate LDs to the same extent. Upregulation of STAT6, GATA3, PCG1 and PPARγ in response to M2-polarizing stimuli (e.g., IL-4, IL-13, TGF-β, IL-10) induces these metabolic adaptations [111–113]. Indeed, the glycolytic rate in M2 macrophages is reduced by the expression of the glycolytic enzyme 6-phosphofructo-2-kinase B1 (PFKFB1) that promotes a reverse step in glycolysis [114]. Instead, M2 macrophages rely more heavily on FAO to fuel OXPHOS and source lipids through scavenger receptor cluster of differentiation 36 (CD36) and lysosomal lipolysis genes [115,116]. Coupled with the activated transcriptional factor LXR in M2 macrophages, the breakdown of lipids for metabolic consumption and cholesterol efflux is favored, preventing the accumulation of LDs as observed in the M1 polarized state [115].
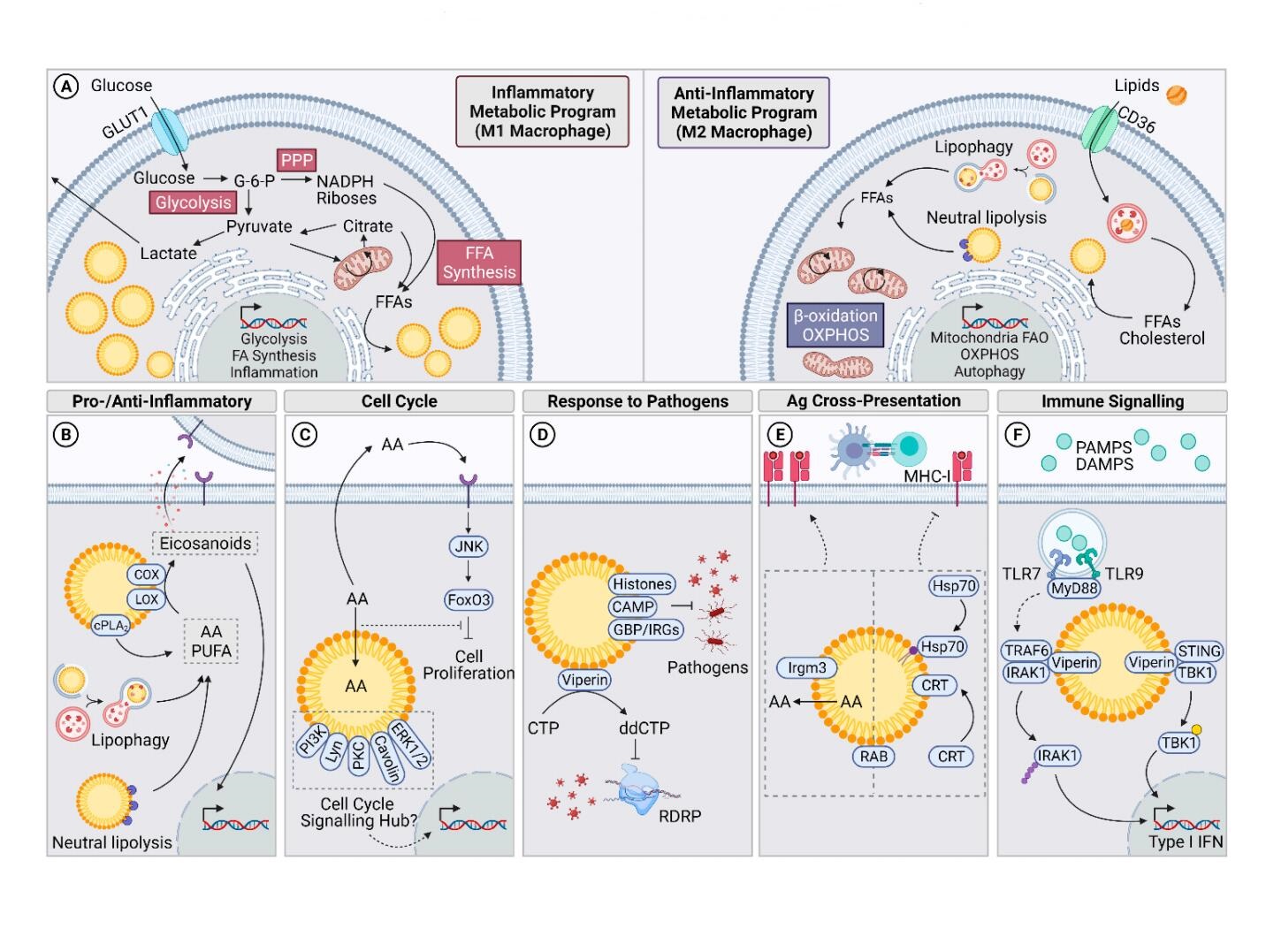 Figure 1. Roles and Regulation of LDs in Activated Immune cells. (A) Metabolic adaptations in cells undergoing inflammatory and anti-inflammatory programs such as M1 and M2 macrophage polarization. Inflammatory cells are highly glycolytic compared to anti-inflammatory cells that rely on oxidative programs. (B) LDs harbour the machinery and store lipids (AA, PUFA) required to produce eicosanoids. (C) LDs may have a role in driving or regulating the cell cycle. PI2K, LYN, PKC, Caveolins and ERK1/2 in the LD proteome suggests that LDs act as cell cycle signalling hubs. Sequestration of anti-proliferative AA may also drive the cell cycle. (D) LDs containing histones, CAMP and/or GBPs and IRGs have direct anti-pathogenic roles. Another LD-resident protein, viperin, catalyses in the production of ddCTP which terminates transcription by RDRP. (E) Irgm3 on LDs enhances antigen cross-presentation on MHC-I. LDs may also regulate this process by sequestration of Hsp70 (recruited by oxidised lipids), calreticulin and RAB proteins. However, this could also serve as a signalling hub to facilitate antigen cross-presentation. (F) Signalling from endosomal TLR7 and TLR9, and STING, is facilitated by LD-resident viperin. Abbreviations: pentose phosphate pathway (PPP), free fatty acid (FFA), fatty acid oxidation (FAO), oxidative phosphorylation (OXPHOS), arachidonic acid (AA), polyunsaturated fatty acids (PUFA), RNA-dependant RNA polymerase (RDRP), calreticulin (CRT).
Figure 1. Roles and Regulation of LDs in Activated Immune cells. (A) Metabolic adaptations in cells undergoing inflammatory and anti-inflammatory programs such as M1 and M2 macrophage polarization. Inflammatory cells are highly glycolytic compared to anti-inflammatory cells that rely on oxidative programs. (B) LDs harbour the machinery and store lipids (AA, PUFA) required to produce eicosanoids. (C) LDs may have a role in driving or regulating the cell cycle. PI2K, LYN, PKC, Caveolins and ERK1/2 in the LD proteome suggests that LDs act as cell cycle signalling hubs. Sequestration of anti-proliferative AA may also drive the cell cycle. (D) LDs containing histones, CAMP and/or GBPs and IRGs have direct anti-pathogenic roles. Another LD-resident protein, viperin, catalyses in the production of ddCTP which terminates transcription by RDRP. (E) Irgm3 on LDs enhances antigen cross-presentation on MHC-I. LDs may also regulate this process by sequestration of Hsp70 (recruited by oxidised lipids), calreticulin and RAB proteins. However, this could also serve as a signalling hub to facilitate antigen cross-presentation. (F) Signalling from endosomal TLR7 and TLR9, and STING, is facilitated by LD-resident viperin. Abbreviations: pentose phosphate pathway (PPP), free fatty acid (FFA), fatty acid oxidation (FAO), oxidative phosphorylation (OXPHOS), arachidonic acid (AA), polyunsaturated fatty acids (PUFA), RNA-dependant RNA polymerase (RDRP), calreticulin (CRT).
These general principles here exemplified in macrophages are replicated in cells of both the myeloid and lymphoid lineage. However, this represents an oversimplification that does not do justice to the complexity of metabolic regulation and influence of the cellular microenvironment. For example, while LDs accumulate in cytotoxic T cells cultured in medium containing optimal glucose, glucose deprivation forces these cells to access fatty acids through lipophagy for FAO, impairing IFNγ production [117]. Furthermore, while immune activation directs metabolic fluxes and LD accumulation, the reverse is also implied. As such, a direct role was attributed to LD formation in conferring the functions of myeloid-derived suppressor cells, independent of other metabolic effects [118]. Impairments in LD turnover mechanisms such as autophagy was shown to promote pro-inflammatory phenotype in murine macrophages [116] and impair neutrophil differentiation [119]. Similarly, LD accumulation fuels the proliferation and inflammatory functions of ILC2 during chronic allergen-driven activation [120]. Although speculative, LDs may play a role in T cell exhaustion [121]. While this highlights the roles of LDs in supporting immune cell function, there remains a complex interplay between metabolic adaptations, the microenvironment and immunogenicity. Altogether, this underlines a multilayered and dynamic response where LDs both result from and support immune activation.
Cells undergoing proliferative events require an increased supply in lipids to both feed regulatory pathways and synthesize new membranes, resulting in the tight regulation of lipidome composition and localization [122]. It is therefore unsurprising that LDs have been observed in many cell types undergoing proliferation. In the absence of other growth factors, exposure of macrophages to various species of phospholipids results in the accumulation of triglyceride-rich LDs and cell proliferation [123]. Moreover, reducing LD formation in murine ILC2 cells through DGAT1 inhibition suppresses the proliferation of these cells [120]. Studies in yeast, hepatocytes, and cancer cells have begun to delineate the role of LDs in cell cycle progression [124–128], suggesting LD formation as a requirement for progression through the G1 phase [126,129,130]. A recent report in non-transformed mammalian cell lines furthermore described the coordination of LD localization and density during the cell cycle [126]. However, the regulation and significance of these events remains elusive [126].
It remains unclear whether the role of LDs is limited to supporting membrane expansion and energy expenditure related to cellular division [130], or whether they actively engage in signalling pathways to drive or regulate cell proliferation (Figure 1C). Various findings substantiate the later idea. Importantly, LDs are the site for eicosanoid and other lipid mediator production [74,131–134], which act in several signalling capacities to regulate the proliferation of immune cells [135–137]. Moreover, by sequestering arachidonic acid (AA) in activated immune cells [138–140], LDs may serve a pro-proliferative role. Indeed, AA was shown to induce cell cycle arrest at the S phase in the RAW264.7 murine macrophage cell line through JNK and FoxO1/O3a signalling, altogether resulting in CyclinD and CDK4 inhibition [141]. Interestingly, the interplay between LDs and FoxO3 signalling was also documented in a colon cancer model, where the pro-proliferative accumulation of LDs was coupled with decreased FoxO3 expression [142]. The localisation of PI3K, ERK1, EKR2, caveolin, Lyn kinase and PKC in cells of both lymphoid and myeloid lineage further suggests that LDs may function as a signalling hub to integrate proliferative pathways in immune cells [63,139,143–145].
With the abovementioned interactions between LDs and the cell cycle, it is interesting to speculate as to how LDs may be involved in senescence, a state where cellular proliferation is arrested while remaining metabolically active [146]. Senescent cells are characterised by the secretion of an array of pro-inflammatory cytokines that are grouped under the term “senescence-associated secretory phenotype” (SASP). Interestingly, an increase in cytosolic LDs is observed in senescent cells of various tissue origins, hypothesized to serve as a protective mechanism against oxidative stress [147–150]. The presence of LDs in non-replicative cells contrasts the idea that LD accumulation directly drives cell cycle progression. Still, this may provide more value to a possible role for LDs as signalling hubs integrating both pro- and anti-proliferation signals.
LDs have an intriguing role in the synthesis and storage of bioactive lipids. Indeed, both the required machinery and substrates to produce pro-inflammatory and pro-resolving mediators have been identified on LDs in activated immune cells [74,131–134]. LDs also serve to regulate the availability of signalling molecules, through the regulated sequestration and release of intermediates generated during TG synthesis and breakdown, which can all act in a signalling capacity [100,151–153]. By protecting against lipotoxicity, LDs additionally buffer FFA-mediated inflammation and stress responses [154]. Consequently, LDs orchestrate cellular programs that integrate immune, metabolic, and inflammatory responses tailored to the activating stimuli [61,100,134,155–157].
Eicosanoids are lipid mediators generally associated with pro-inflammatory roles, as well as with the migration, proliferation, and activation of immune cells [134,158,159]. In recent years, their roles have been nuanced with the identification of new functions in dampening the inflammatory and immune response [155,160]. Stored in esterified form, the eicosanoid precursor AA is generated from the hydrolysis of glycerophospholipids by phospholipase A2 (PLA2) family enzymes [161,162]. With more than 30 members, PLA2 enzymes are classically categorized as secreted (sPLA2), intracellular calcium-independent (iPLA2) or cytosolic calcium-dependent (cPLA2). Of these, cPLA2α is the main contributor to the hydrolysis of membrane phospholipids containing AA [161]. In stimulated immune cells, cPLA2α translocates to the surface of LDs [132,162–164]. This interestingly coincides with the mobilization of AA from the phospholipid to the triglyceride pools during immune cell activation, thus enriching LDs in AA [138–140]. Recent studies furthermore implicate neutral lipolysis (ATGL [165,166], MAGL [167,168]) and lysosomal lipolysis (LAL) [169,170] in sourcing AA for eicosanoid production. Following hydrolysis by cPLA2α, AA or related polyunsaturated fatty acids can undergo oxidative modifications to form eicosanoids, mediated by the cyclooxygenase (COX), lipoxygenase (LOX) and cytochrome P450 pathways [159]. The localization of these enzymes within LDs in activated immune cells, as well as the in situ identification of eicosanoids such as PGE2 further strengthens the role of LDs in the synthesis of these bioactive lipids (Figure 1B) [74,131–134]. The documented roles of cPLA2α and other cPLA2 in the biogenesis of LDs additionally fortifies the link between LDs and the eicosanoid-producing machinery [162,171,172]. Recently, the synthesis of TGs and LD assembly in response to M1-polarizing stimuli was found to be essential to optimize the inflammatory response in macrophages [110]. Inhibition of DGAT1-mediated TG synthesis and LD biogenesis impaired IL-1β, IL-6 and PGE2 synthesis, as well as phagocytic capacity of the cells [110].
Yet, there remain many open questions as to the exact contribution of LDs in regulating the availability of AA. More specifically, the association of AA with various lipid species within LDs and other cellular lipids raises questions about the significance of LD-associated AA. For a long time, LDs have been known to store AA in both their neutral core and at their phospholipid monolayer [139,173]. While cPLA2α mediates the release of AA from membrane phospholipids, how the AA-enriched TG core of LDs in activated immune cells may supply AA for eicosanoid production remains unclear due to the LD core accessibility and the phospholipid-specific hydrolase function of cPLA2α [138–140]. Recent studies in macrophages have demonstrated that the neutral lipid AA pool may only contribute modestly to the synthesis of bioactive lipids [140]. Also, stimuli-dependent remodeling of AA specie-specific associations with the phospholipid pool may serve as a mechanism to regulate the activity of cPLA2α and hints at the importance of this pool in modulating bioactive lipid synthesis [89,174,175]. If organelle-specific AA-phospholipid association is regulated, how LD-specific phospholipid AA remodeling regulates immunity is unclear. On one hand, LD sequestration of AA modulates inflammatory responses by limiting its bioavailability [100,176]. On the other hand, through an undescribed mechanism, TG enrichment may replenish membrane phospholipid AA and drive eicosanoid production at the LD [100,134,177]. However, the ability of cPLA2 to release AA from phospholipids at the LD monolayer was not directly confirmed. Adding to this complexity, enrichment of lipid fractions with other fatty acids that interact with the same machinery as AA in non-redundant manners, such as adrenic acid, was observed [89,176,178]. Altogether, this paints a picture where LDs play a central role in the production and modulation of eicosanoids and other bioactive lipids.
LDs SUPPORT HOST IMMUNITYLDs have recently come to be appreciated as frontline responders against invaders, harboring antimicrobial proteins and forming signalling hubs for immune-related pathways (Figure 1D). A direct antimicrobial role for LDs was recently shown with identification of cathelicidin (CAMP) in the LD proteome of human monocyte-derived macrophages [179]. LD accumulation in macrophages effectively reduced the intracellular survival of E. Coli, and this antibacterial effect was abrogated in CAMP-silenced macrophages [179]. Moreover, the histones H2A, H2B and H4 were identified in the LD proteome of LPS-treated murine hepatocytes and the U937 human monocyte cell line [179,180]. Studies in Drosophila have described the storage of histones on LDs both as a mechanism to regulate the availability of these important DNA-associated proteins [181] and as an antibacterial strategy [57,61,182]. Histone-mediated antimicrobial effects have since been described against various bacterial, viral, parasitic and fungal pathogens [183].
The role of the interferon response in many pathogenic infections is undeniable, serving to mediate intercellular communication and induce antimicrobial functions [51–57]. Mechanistically, sensing of pathogens by PRRs triggers the production of interferons, which subsequently signal in autocrine, paracrine, and endocrine manners to activate interferon stimulated genes (ISGs). Several ISG products have been identified in the LD proteome and may exert direct antipathogenic functions against invaders that exploit LDs as part of their replicative cycle and evasion mechanisms [53,55,179]. Viperin is perhaps one of the most discussed ISG product that localises at the surface of LDs and has been the subject of recent reviews detailing its antiviral roles [184,185]. Its expression is induced both by initial PRR pathogen sensing and upon interferon feedback signalling [184,185]. Viperin interacts with a wide range of host and viral proteins to inhibit viral entry, replication, assembly and budding [184,185]. Moreover, viperin contributes to signal transduction from the T cell receptor (TCR), TLR7, TLR9, and Stimulator of Interferon Genes (STING) pathways (Figure 1F) [186–189]. Pertaining to its role in TLR7 and TLR9 signalling, viperin co-localizes with IRAK1 and TRAF6 on LDs, serving as a scaffold to facilitate K63-linked ubiquitination of IRAK1 by TRAF6 [188,189]. IRAK1 subsequently signals downstream to mediate the type I interferon response [188–190]. MyD88 was suggested to relay TLR9 signalling from endosomes to LDs, as it localizes to LDs at moderate frequency [189]. A similar role for LD-localized viperin in STING signalling was recently described, where in response to dsDNA, TBK1 and STING co-localized with viperin to activate TBK1 and, consequently, interferon production [186]. These findings are further substantiated by recent studies highlighting that LDs are required to enhance the interferon response [67,190]. Moreover, a catalytic role for viperin in the formation of the viral polymerase inhibitor ddhCTP was shown (Figure 1D). CMPK2, an antiviral gene co-transcribed with viperin upon interferon signalling [184,191,192], phosphorylates cytidine diphosphate (CDP) to produce cytidine triphosphate (CTP) [193]. Viperin can then initiate the radical-mediated dehydration of CTP into 3ʼ-deoxy-3ʼ,4ʼ-didehydro-cytidine triphosphate (ddhCTP), which can be incorporated by the viral RNA-dependant RNA polymerase and abort the transcription of viral RNA [194,195]. Interestingly, the association of viperin with IRAK1 and TRAF6, or with STING and TBK1, increased the production of ddhCTP, indicative that binding of viperin to these signalling proteins may regulate this protein’s enzymatic activity [188]. This also led to increased viperin degradation, which may serve as a regulatory mechanism to dampen antiviral responses [186,188].
Using hepatic LDs as a proof of concept that mammalian LDs participate in innate immunity, Bosch et al. analyzed the LD proteome in response to LPS treatment, identifying the differential expression of 689 proteins on these organelles, with 317 enriched and 372 reduced following treatment [179]. These changes in the LD proteome were correlated to antibacterial activity, metabolic adaptations, and physical and functional uncoupling of LDs from mitochondria. As such, proteins integrating innate immune functions were found upregulated, in contrast with the observed downregulation in proteins involved in OXPHOS, NAFLD, the TCA cycle and fatty acid catabolism. Importantly, stimulation with LPS, TNFα and IFNγ differentially regulated the LD proteome, highlighting a requirement for stimuli-specific LD remodeling in integrating appropriate immune responses.
Several immune-related GTPases (IRGs), including GVIN, IFGGA1, IFGGB55, and IFI35, were also found clustered with PLIN2 [179]. IGTP, IIGP1, TGTP1 and IFI47 were additionally identified directly associated with LPS-induced LDs, further solidifying the role of these organelles in coordinating the innate immune response [179]. The upregulation and shuttling of IRGs and guanylate-binding proteins (GBPs) to LDs has been reported in many instances of pathogenic infection (Figure 1D) [179,196]. The antipathogenic functions mediated by these proteins were previously been summarized [197] and includes the regulation of autophagy [198–202], suggesting a role for IRGs in regulating LD degradation [200,201,203]. Interestingly, a function for IRG IFGGA2 in inducing lipophagy was recently observed and proposed to prevent excessive LD accumulation in a model of fatty liver disease [203]. This same protein was found upregulated on LPS-induced LDs [179]. In contrast, a role for IRG member Irgm3 in LD formation was identified in dendritic cells, in which Irgm3 deficiency abrogates LD accumulation [204]. How IRGs regulate the biogenesis and catabolism of LDs to regulate lipid metabolism in immune cells, target pathogen-seized LDs for degradation, directly exert antipathogenic functions and prevent lipid accumulation remains to be further investigated. Reconfiguration of the LD proteome in a stimuli-specific manner and the scaffolding of immune proteins on LDs highlights their role in coordinating immunometabolic responses.
ROLE OF LDs IN ANTIGEN CROSS-PRESENTATIONWhile debated, LDs may serve a role in antigen cross-presentation (Figure 1E). PLIN2-associated Irgm3 on LDs was shown to regulate antigen cross-presentation, enabling dendritic cells to present exogenous antigens on MHC-I molecules at the cell surface and mediate the activation of cytotoxic CD8+ T cells [204–206]. In PLIN2- or Irgm3-deficient dendritic cells stimulated with IFNγ, antigen cross-presentation as measured by CD8+ T cell activation is impaired [204]. Moreover, hepatic dendritic cell populations with elevated lipid were shown to readily activate T cells, NK cells and NKT cells, and blocking fatty acid synthesis reduced their immunogenicity [207]. Conversely, hepatic dendritic cells with reduced lipid content stimulated regulatory T cells and tolerogenic responses. Recently, a saponin-based adjuvant used to enhance T cell responses was shown to promote antigen cross-presentation by increasing dendritic cell LD biogenesis [208]. Although the mechanisms underlying the positive contribution of LDs to antigen cross-presentation remain elusive, a role for Irgm3 in antigen translocation to the cytosol was hypothesized [206]. It was also suggested that AA release from LDs may activate NADPH oxidase to enhance the acidification of endosomal compartments required for antigen proteolysis and cross-presentation [205,209,210].
In striking contrast, the accumulation of LDs has also been associated with impaired antigen cross-presentation. Of note, these studies mainly investigated the immunogenicity of dendritic cells in tumor microenvironments [211]. Mechanistically, accumulation of lipids that have undergone oxidative modification within cytosolic LDs results in recruitment and trapping of the major stress-induced peptide chaperone heat shock protein 70 (Hsp70) [212,213]. As Hsp70 participates in the trafficking of peptide-loaded MHC-I molecules to the plasma membrane [214], its sequestration within LDs hinders antigen cross-presentation in dendritic cells [212]. Interestingly, another mediator of antigen cross-presentation, calreticulin [215,216], is sequestered in LDs in colon cancer cells treated with chemotherapeutics [217]. This leads to decreased engulfment of dying cancer cells by dendritic cells due to reduced exposure of calreticulin at the cell surface that acts as an “eat-me” signal. While calreticulin sequestration in LDs by antigen presenting cells has not been shown, the concept that Hsp70 could be sequestered in LDs in response to activating stimuli is supported by the proteomic analyses of LPS-induced LDs in hepatocytes [179]. Similarly, several RAB GTPases are enriched on LDs following LPS stimulation [179], some of which have been involved in antigen cross-presentation (e.g., Rab14 [218], Rab34 [219]) [220]. If calreticulin or RAB protein sequestration in LDs could coordinate immunity through regulating antigen cross-presentation is an interesting concept that should be further explored. Altogether, the contrasting functions of LDs in antigen cross-presentation once again highlights the dynamic nature of LDs in response to environmental and metabolic cues. The known implications of antigen cross-presentation in mounting cytotoxic T cell responses in many pathogenic infections and neoplastic diseases warrants further studies investigating the role of LDs in antigen cross-presentation.
With their network of organelle interactions, LDs are at the crossroads of several metabolic pathways that drive immune responses [18,51,56,57,221,222]. LDs make contacts with several organelles, including the nucleus, mitochondria, peroxisomes, the ER, lysosomes and autophagosomes [19]. In nuclear crosstalk, regulation of the nuclear factor of activated T cells (NFAT5) by the CIDE family protein Fsp27 is emerging as a critical regulator of immune cell activation, especially T cells and macrophages (Figure 2A) [223]. LD-residing Fsp27 can interact with NFAT5, sequestering it to the cytoplasm and preventing its nuclear translocation [224], but the immunomodulatory role of this interaction remains unclear. PLIN5 also coordinates gene expression, via extranuclear activation of regulatory pathways and by translocation to the nucleus [225]. As such, in lipid-stressed pancreatic β cells, PLIN5 induces PI3K/Akt/ERK signalling, resulting in the nuclear translocation of Nrf2 and transcription of antioxidant enzymes GCLC and HO-1 [225]. In response to catecholamine-induced cAMP/PKA activation, PLIN5 phosphorylation and its translocation to the nucleus ensues, where PLIN5 associates with SIRT1 and PGC-1α to mediate gene transcription [226]. This regulates metabolic pathways that increase fatty acid catabolism and mitochondrial respiration and correlate with the inhibitory role of PLIN5 on LD lipolysis [227,228]. Translocation of PLIN5 to the nucleus hence stimulates LD lipolysis and coordinates the supply of fatty acids for FAO [226]. Moreover, PLIN5 participates in LD-mitochondrial interactions that may facilitate FAO metabolism through physical proximity of the two organelles. Expression of PLIN5 in macrophages is upregulated in response to oxidised low-density lipoprotein (oxLDL) to modulate inflammatory responses, whereas PLIN5 knockdown in macrophages increases inflammatory gene expression [229]. Finally, an additional layer of inter-regulation between the nucleus and LDs pertains to the CCTα enzyme. In Drosophila and murine macrophages, CCTα shuttles between the nucleus and cytoplasm. Upon exposure to the fatty acid oleate, CCTα is targeted to the surface of LDs, activating the synthesis of PC that results in LD expansion [6]. As LDs residing in the nucleus also expand under the action of the CCTα enzyme [37,38,48], it will be interesting to see if further investigation reveals a role for the translocation of CCTα in regulating nuclear versus cytoplasmic LD formation.
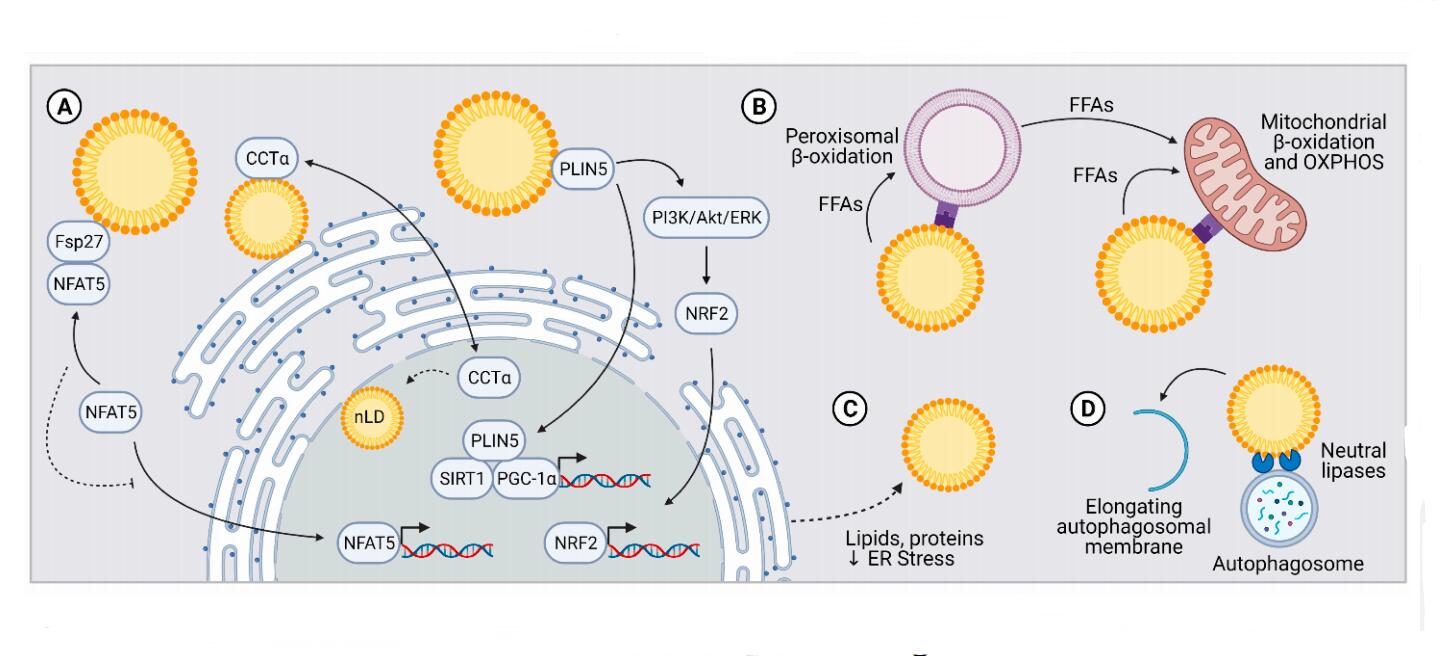 Figure 2. LDs Regulate Immune Responses through Inter-Organelle Interactions. (A) LDs interact with the nucleus by regulating nuclear translocation of transcription factors (NFAT5, PLIN5) or lipid synthesis enzymes (CCTα). CCTα regulates LD biogenesis in the cytosol vs. nucleus. (B) The direct transfer of FAs from LDs to mitochondria or via a peroxisome intermediate helps coordinate oxidative metabolic programs. (C) Transfer of lipids and proteins from the ER to LDs mitigates ER stress and dampens immune activation driven by ER stress. (D) LD interactions with autophagosomal membranes serve as a scaffold for neutral lipases. LDs also promote autophagosome membrane elongation. Abbreviations: nLD (nuclear LD), free fatty acid (FFA), oxidative phosphorylation (OXPHOS).
Figure 2. LDs Regulate Immune Responses through Inter-Organelle Interactions. (A) LDs interact with the nucleus by regulating nuclear translocation of transcription factors (NFAT5, PLIN5) or lipid synthesis enzymes (CCTα). CCTα regulates LD biogenesis in the cytosol vs. nucleus. (B) The direct transfer of FAs from LDs to mitochondria or via a peroxisome intermediate helps coordinate oxidative metabolic programs. (C) Transfer of lipids and proteins from the ER to LDs mitigates ER stress and dampens immune activation driven by ER stress. (D) LD interactions with autophagosomal membranes serve as a scaffold for neutral lipases. LDs also promote autophagosome membrane elongation. Abbreviations: nLD (nuclear LD), free fatty acid (FFA), oxidative phosphorylation (OXPHOS).
The regulation of FAO by LDs elegantly intersects at the nucleus and mitochondrial junction (Figure 2B). In many tissues CPT-1, a major regulator of FFA trafficking to mitochondria, is under transcriptional regulation by SIRT1/PGC-1α [230]. These transcription factors are enhanced by PLIN5 [226]. Nuclear translocation of LD-associated PLIN5 therefore enhances both LD lipolysis and uptake of FAs by mitochondria, further elevating the role of LDs in regulating metabolic pathways that drive immune cell functions. LDs also coordinate lipid metabolism through direct physical interactions with mitochondria. TGs stored in LDs can be released through regulated lipophagy and neutral lipolysis, and subsequently used by mitochondria to fuel the FAO and OXPHOS. Under conditions of starvation, LDs associate with mitochondria, conferring an advantageous proximity to support bioenergetic pathways [231–240]. In mouse embryonic fibroblasts, LD-mitochondria interactions depend on lipolysis, autophagy, and mitochondrial fusion, ultimately supplying mitochondria with FFAs for β-oxidation [234]. However, contradictory findings that mitochondria surrounding LDs prevent lipolysis and mediate LD expansion in brown adipose tissue highlight the importance of tissue- and context-specific characterization of these interactions [241]. Mechanistically, notable protein candidates that mediate LD-mitochondria interactions include PLIN1 [242], PLIN5 [243,244], MFN2 [242,245], MIGA2 [246], SNAP23 [247] and RAB32 [236,248]. Identification of other LD-mitochondria contact mediators and characterization of their role and regulation may provide further insight into these contradictory functions associated with LD-mitochondria contacts.
LD-mitochondria contacts are intriguingly understudied in immune cells, raising many open questions as to the impact of both organelles in immune functions. The downregulation of LD-resident PLIN5 in response to LPS challenge was demonstrated in hepatocytes, resulting in decreased β-oxidation and ketogenesis due to the uncoupling of mitochondria and LDs [179]. This uncoupling increases the availability of LDs for antimicrobial interactions and remodels metabolic fluxes to sustain immune activation [179]. Conversely, PLIN5 overexpression in human THP-1 macrophages increased LD-mitochondria contacts and reduced antibacterial function [179]. The reliance of antibacterial, proinflammatory M1 macrophages on glycolysis as compared to the preferential use of FAO and OXPHOS in anti-inflammatory M2 macrophages supports the rationale for LD-mitochondria uncoupling in response to M1-polarizing stimuli such as LPS [101]. Identification of an inflammation-dampening role for PLIN5 in oxLDL-treated RAW264.7 murine macrophages further supports the immunomodulatory role of LD-mitochondria coupling [229]. If these contacts drive and/or result from the rewired metabolic fluxes in immune cells, and if increasing or decreasing these contacts skews immune cell function remains to be elucidated.
In mammalian cells, peroxisomes and mitochondria share the ability to perform β-oxidation. In mitochondria, long-chain fatty acids are processed for energy production, whereas in peroxisomes, very-long chain and branched fatty acids are used for the biosynthesis of specific fatty acids and H2O2 production [249]. Fatty acids generated by peroxisomal β-oxidation may thereafter supply mitochondrial energy metabolism (Figure 2B) [249]. Interestingly, peroxisomal contribution to lipid metabolism in immune cells of both lymphoid and myeloid origin correlates with cell proliferation and immune activation and function [250,251]. Notably, peroxisome activation in macrophages reduces COX-2 expression and pro-inflammatory cytokines in response to LPS [252]. In contrast, knockdown of β-oxidation-associated peroxisomal genes enhances inflammatory mediator production [252]. LDs can directly interact with peroxisomes to promote fatty acid trafficking between the two organelles [237,253,254]. These contacts were recently found to be mediated via interactions between the LD-resident M1 Spastin and peroxisome-resident ABCD1 [255], although their role in regulating immune function is unknown. Given that LDs and peroxisomes share mutual functions in the production of pro- and anti-inflammatory mediators [250], it is tempting to speculate that their coupling results in the synergistic execution of these roles.
LDs are known to accumulate in response to ER stress [256]. The impacts of ER stress and the unfolded protein response (UPR) on immune cells have been well described, but how LD-ER interactions regulate these processes remain elusive [240,257,258]. Studies linking the UPR, LDs and immunity are lacking. In yeast, LDs can attenuate ER stress through the removal of lipids and misfolded proteins [259], but whether this process is replicated in immune cells to mitigate immune activation remains to be investigated (Figure 2C). Finally, while CMA and macroautophagy have established roles in lipophagy, microautophagy of LDs has only recently been linked to mammalian lipophagy [22,25]. Indeed, contacts between lysosomes and LDs resembling the process of microautophagy have been described in hepatocytes and macrophages [22,25]. Much remains to be understood about mammalian microlipophagy, but further elucidation of its mechanisms may yield interesting discoveries in immunometabolism. For example, distinct LD lipid compositions may contribute to preferential targeting of LDs to macro or microautophagy [260], which may relate to lipidome remodeling of LDs during immune activation. Whether microlipophagy is differentially regulated during immune activation, and whether it may contribute to metabolic adaptations will be fascinating to uncover. In addition to lipophagy, interesting interactions have been described between the autophagy machinery and LDs (Figure 2D). In yeast and mammalian cells, LDs may serve as a lipid source for autophagosomal membranes elongation in certain conditions [261–263]. While this direct contribution was shown to be expendable, the mitigation of ER stress by LDs may nevertheless contribute to this process by conserving ER homeostasis and function [264]. Autophagosomes may also serve a scaffolding role during LD cytosolic lipolysis [26,265], yet how this is impacted by immune metabolic programs is currently unknown.
LDs IN SARS-CoV-2 SURVIVALDespite the antiviral properties of LDs, several viruses can leverage these organelles to support to their life cycle, including viral entry, translation, assembly of new particles and release [56,61,266,267]. This striking contrast to their antiviral properties has been identified as part of the pathogenic strategy employed by many positive-sense single-stranded RNA viruses ((+)ssRNA), including non-exhaustively reoviruses, polioviruses, flaviviruses, and coronaviruses [61]. Given the immense volume of new information that is published daily on coronaviruses as a result of the ongoing global pandemic, we herein focus our attention on Sars-CoV-2, the virus responsible for Covid-19 disease.
The known ability of some Coronaviradae family members to remodel and utilise lipid metabolism has prompted the investigation of LD hijacking by SARS-CoV-2 [268–270]. SARS-CoV-2 is an enveloped, (+)ssRNA virus able to infect mammalian cells through its interaction with the ACE2 receptor [271,272]. Once internalised, the viral particles shed their envelope and immediately initiate the translation of their RNA genome to generate the viral replication and transcription complex. This complex mediates the synthesis of viral RNA through the RNA-dependant RNA polymerase (RDRP), providing the necessary genomic and structural material for the assembly of new virions. These virions finally exit the cell through exocytosis, completing the virus cycle.
Cytokine storm, a condition defined by the hyperactivation of immune cells causing an exacerbated release of inflammatory cytokines, is associated with severe Covid-19 disease [273,274]. This heightened inflammatory phenotype is accompanied by significant dyslipidemia and immune dysfunction [269,275–278]. Importantly, pro-inflammatory monocyte-derived macrophages present in high quantity in the bronchoalveolar lavage fluid of severe Covid-19 patients [279]. Through a feedback loop involving lung-resident macrophage death and monocyte and T cell recruitment, alveolar macrophages perpetuate the inflammatory response and immune activation to SARS-CoV-2 [280]. This underlines a role for macrophages in the pathogenesis and severity of Covid-19 [274,281]. Monocytes infected in vitro with SARS-CoV-2 upregulate CD36, SREBP-1, PPARγ, and DGAT-1, increasing lipid uptake and LD accumulation [282]. This benefits the SARS-CoV-2 virus that utilizes LDs as replication platforms [282]. Preventing LD accumulation with DGAT-1 inhibitors reduces viral replication and the release of inflammatory mediators by monocytes, further supporting a role for LDs in SARS-CoV-2 pathogenesis [282].
The role of LDs in Covid-19 disease may also provide insight into the heightened rate of hospitalization observed in individuals that are aged, obese and/or suffer from metabolic disorders [283,284]. Hyperglycemic conditions enhance glucose uptake and aerobic glycolysis of SARS-CoV-2-infected monocytes in vitro, concurrently augmenting viral load, ACE2 and IL-1β expression [285]. Viral load from in vitro infected monocytes is increased in cells isolated from obese or diabetic individuals as compared to that of healthy controls, suggesting a role for environmental conditioning [285]. Moreover, murine bone-marrow derived macrophages extracted from obese mice present with decreased basal autophagy and enhanced pro-inflammatory M1 polarization in response to IFN-γ or LPS stimulation [116]. Polarization to M1 proinflammatory macrophages favors metabolic programs enhancing LD accumulation, which may correlate with the heightened SARS-CoV-2 viral load in hyperglycemic individuals. Similarly, the accumulation of senescent cells associated with the aging process may contribute to Covid-19 disease severity [286,287]. Senescence-induced metabolic alternations including LD accumulation, decreased autophagy and the development of a chronic inflammatory phenotype may contribute to pathogenesis by creating an intracellular environment favorable for SARS-CoV-2 replication [146–150].
Several therapeutic approaches involving cellular metabolism have been proposed, with much interest in the modulation lipid metabolism [270,278,288–291]. Notably, many autophagy-targeting compounds are under investigation [291]. However, the importance of autophagy in several immune processes, and our limited understanding of how SARS-CoV-2 interacts with this machinery has raised concerns regarding its modulation [292–294]. Importantly, our review provides further rationale to air on the side of caution regarding autophagy modulation. Indeed, the interplay between LD-specific autophagy, termed lipophagy, and SARS-CoV-2 in disease pathogenesis and antiviral immunity has not yet been characterised. As such, inhibiting lipophagy could increase the accumulation of LDs used by SARS-CoV-2 in its replication cycle [282]. In uninfected immune cells, reducing LD density by enhancing lipophagy could impair the LD-mediated antiviral effects and skew immune function [57,61,64,100,190]. In contrast, promoting LD accumulation through lipophagy inhibition could further potentiate the production of inflammatory mediators and the cytokine storm [58,61,100,282].
LDs IN STERILE INFLAMMATIONAtherosclerosis is a sterile inflammatory disease driven by the accumulation of LD-enriched foam cells in the vascular wall [295]. In atherosclerosis, monocytes are recruited to the arterial wall in response to a buildup of cholesterol-enriched lipoproteins [296]. Within the subendothelial space of the vascular wall, monocytes differentiate into macrophages that function to phagocytose excess modified lipoproteins, leading to macrophage foam cell formation [295]. Mounting evidence indicates that, in parallel, intimal vascular smooth muscle cells (VSMCs) originating from the medial artery layer can also become lipid-laden and develop into foam cells [297–301]. Given that an accurate inventory of LD proteins is crucial to understanding LD functions [302], the protein composition of cytoplasmic LDs in macrophages has been extensively analyzed by proteomics [22,179,303], whereas the LD proteome of VSMC foam cells has not yet. Proteins previously identified on macrophage LDs include structural proteins of the PLIN family (e.g., PLIN2, PLIN3), TG and CE lipolysis machinery (e.g., HSL, patatin like phospholipase domain containing 2 [PNPLA2], CES1, lipid droplet hydrolase [LDAH]), ubiquitination factors (e.g., ancient ubiquitous protein 1 [AUP1], ubiquitin-conjugating enzyme E2 G2 [UBE2G2]) and RAB proteins (e.g., RAB5A, RAB7A, RAB18) [302].
In a study aimed at identifying novel genes that modulate lipid storage in macrophages, Mejhert et al. uncovered that Max-like protein X (MLX) transcription factors, key regulators of multiple metabolic adaptations to glucose, bind to LDs, and that these interactions regulate metabolic gene expression [303]. Binding of MLX transcription factors to the surface of LDs blunted their transcriptional activity in response to glucose, ensuring an appropriate cell response to fluctuating glucose concentrations depending on whether LDs were abundant, signaling that abundant cellular energy stores are present [303]. More recently, novel candidate lipophagy selectivity factors were identified on LDs by mass spectrometry, including autophagy regulators LAMTOR5, HMGB1 and HMGB2, RAB8 and DAP, the cholesterol efflux regulator SCARB2 [22]. The expression of several LD-associated lipophagy factors identified in this study were observed to be dysregulated in macrophages during atherosclerosis development, suggesting that alterations in lipophagy flux may contribute to atherogenesis [22]. Given that that altering cellular metabolism can regulate macrophage M1 and M2 inflammatory polarization within atherosclerotic plaques, it is tempting to speculate that macrophage lipophagy plays a key role in regulating LD hydrolysis to control the availability of fatty acids to undergo β-oxidation required to sustain M2 effector functions such as tissue repair and inflammation resolution, and that this accounts for some of the observed protection that macrophage autophagy confers against atherosclerosis [304–307]. Intriguingly, both single LDs and clusters of LDs appear to be tagged for lipophagy-mediated catabolism in macrophage foam cells [22], although how the formation of LD clusters might regulate immunometabolism is currently unclear. Future studies are needed to decipher the functional significance of LD clusters, using 3D imaging and image rendering to better detect their presence as they cannot be depicted equivalently in a 2D image [308].
PLIN family members exhibit a tissue-specific expression pattern: PLIN1 is a central regulator of LD storage and breakdown in adipose tissue, PLIN2 and PLIN3 are highly expressed in most tissues, PLIN4 primarily resides in white adipose tissue LD populations distinct from PLIN1, and PLIN5 is expressed in tissues with elevated rates of FAO [309]. Although PLIN1 is not basally highly expressed in macrophages, its expression is upregulated upon foam cell formation, and whole-body and bone marrow PLIN1 deficiency reduces atherosclerosis in Apoe−/− by 39% and 49% respectively [310]. Conversely, two studies observed a protective role for PLIN1 in atherosclerosis, where atherosclerosis was increased in Ldlr−/− mice lacking PLIN1 [311] and PLIN overexpression in macrophages reduced atherosclerosis and skewed macrophages towards an anti-inflammatory phenotype [312]. In turn, PLIN2 is the most highly expressed PLIN in human and mouse macrophage foam cells and promotes a M1 proinflammatory phenotype [313–315]. In line with this, PLIN2 inactivation in Apoe−/− mice impairs LD biogenesis in macrophages and protects against atherosclerosis [315], and inhibition of PLIN2 was proposed as a strategy to counteract metabolic and age-related diseases in which a characteristic feature is the buildup of PLIN2 LDs [309]. Finally, PLIN5 deficiency promotes atherosclerosis progression by increasing inflammation, apoptosis, and oxidative stress [229]. PLIN5 deficiency also alters cardiac lipid metabolism and reduces survival post-myocardial ischemia, highlighting a protective role for PLIN5 in the heart [316]. On balance, these studies show that the role of PLINs in LD biogenesis and turnover and their impact on macrophage inflammation and atherosclerosis is non-negligible. Regulating the expression of PLIN proteins may represent a viable strategy to treat atherosclerosis and other diseases with metabolic dysregulation. To this end, one study proposed an inhibitor of PLIN1 as a potential treatment for obesity, given that PLIN1 degradation or knock-out is associated with leanness [317]. Whether similar inhibitors could be designed to block atherosclerosis-relevant PLINs and the role of LDs in driving inflammatory responses in other myeloid cells known to accumulate LDs in atherosclerosis, such as neutrophils and dendritic cells, remains to be seen.
LDs store polyunsaturated FAs (PUFAs) that are precursors for the synthesis of bioactive lipid mediators, such as eicosanoids and specialized pro-resolving mediators (SPMs) [100]. The later have gained increasing attention in the atherosclerosis research field in recent years, with emerging evidence that resolvins, belonging to the group of SPMs, limit neutrophil infiltration, promote tissue remodeling and homeostasis, and stimulate the clearance of apoptotic and necroptotic cells by macrophages [318–320]. Imbalances between SPMs and leukotrienes (LTs), particularly the pro-resolving resolvin D1 (RvD1) and the pro-inflammatory leukotriene B4 (LTB4), promotes the instability of atherosclerotic plaques [319]. Boosting inflammation resolution in atherosclerosis by promoting SPM biosynthesis therefore holds promise for immunomodulatory treatment of cardiovascular diseases [321], and future studies are needed to determine how LDs tie into regulating SPM and LT biogenesis in this context.
In the central nervous system (CNS), the presence of LDs is minimal and most noticeable in glial cells. Across all species, the brain is comprised of two major classes of cells: neurons and glial cells. Neurons possess electric excitability and are the main component of the nervous system, while glial cells provide structural and metabolic support. Glial cells can be classified as oligodendrocytes, astrocytes, and microglia. Astrocytes are the most abundant CNS glial cells and are closely associated with neuronal synapses to regulate the transmission of electrical impulses in the brain. Oligodendrocytes are the myelinating cells of the CNS and provide insulation to neuronal axons. Finally, microglia are brain-resident macrophages that confer CNS immune defense. As early as 1907, a striking buildup of “adipose saccules” were observed in the post-mortem brain of Alzheimer Disease (AD) patients, closely followed by the observation of LDs accumulating in glia during the normal process of aging [322–327]. In many models of neurodegeneration, excessive glial LD accumulation is now considered a hallmark of disease [328,329]. Neuroinflammatory changes such as microglial activation and the release of pro-inflammatory cytokines have been consistently observed in neurodegenerative diseases and normal aging [323,330]. The accumulation of LDs under these conditions is believed to be a consequence of excess exogenous lipid availability, increased myelin fragmentation or cell stress [331–333]. However, in the CNS, the functional role of excess LDs in glial cells in driving immune responses is not well understood. Interesting correlations can nevertheless be drawn using existing knowledge to reflect on the possible roles played by LDs in brain cells.
While the role and formation of LDs in neurons under physiological conditions remains poorly understood, their role in the context of many neurodegenerative pathologies is unequivocal [328]. Generally, excessive neuronal stimulation leads to the build-up of reactive oxygen species (ROS) and the accumulation of fatty acids via SREBP-mediated lipogenesis (Figure 3A). Peroxidation of accumulated fatty acids, particularly polyunsaturated fatty acids (PUFAs) such as AA and docosahexaenoic acid (DHA), can lead to neuronal damage and eventual neurodegeneration [334,335]. The availability of these lipids is required for activation of signalling and inflammatory pathways. To alleviate this burden, neurons transfer their accumulated lipids to astrocytes via apolipoproteins E and D and subsequently store the lipids as LDs [336]. As such, incorporation into LDs not only protects PUFAs from peroxidation and subsequent neuronal damage, but also limits the synthesis of inflammatory lipid mediators [331]. In glial cells, the integration of DHA in LDs induces their remodelling and increases the prevalence of interactions between LDs and mitochondria, suggesting an increased in FAO metabolism [337]. LD-laden astrocytes also exhibit markedly increased expression of the glial fibrillary acidic protein (GFAP) that is characteristic of astrogliosis, defined as the abnormal increase in the number of astrocytes due to the destruction of nearby neurons from CNS trauma, infection, ischemia, stroke, autoimmune responses, neurodegenerative disease, or obesity-induced hypothalamic inflammation [338]. Interestingly, obesity leads to crosstalk between astrocytic foam cells and microglia, whereby lipid-laden astrocyte-derived inflammatory mediators promote microglia migration and activation to potentiate hypothalamic inflammation in obesity [338]. Whether LDs result from or trigger astrocytic inflammatory responses remains to be determined.
Microglia are brain-resident macrophages that are key players in CNS immunity [339]. Microglia can undergo metabolic reprogramming based on the availability of nutrients or in response to neuroinflammatory cues to effectively maintain their immune function [340]. During inflammation and aging, microglial activation promotes phagocytosis, cytokine release and ROS production [324,341]. This process involves the accumulation of larger LDs in the brain [323,326]. Additionally, microglial cells undergo phenotypic changes to adapt to their changing microenvironment [340]. Previously described microglial phenotypes include the pro-inflammatory M1 phenotype, the immunosuppressive M2 phenotype, dark microglia (inactivated microglial phenotype predominantly associated with pathological states and exhibiting elevated oxidative stress [342]), and disease-associated-microglia (DAM). More recently, LD-accumulating microglia (LDAM) were identified as a unique subset of microglia in aging mice with distinct transcriptional profiles (Figure 3B) [323]. Interestingly, these cells exhibit defective phagocytosis and possess similar transcription profiles to LPS-induced microglia. The LDAM phenotype has also been observed in microglia depleted of lipoprotein lipase (LPL), and was amenable to the addition of peroxisome proliferator-activated receptor (PPAR) agonists to mitigate inflammation [343,344]. LDAM and LPS-induced microglia excessively release pro-inflammatory cytokines, implying a role for LD accumulation in LPS-induced inflammation and aging-induced inflammation.
More recently, prostaglandin E2 (PGE2) was implicated in aging and AD, whereby PGE2 levels increase in myeloid cells of the ageing brain and PGE2 signaling through its EP2 receptor increases glucose sequestration in glycogen, driving maladaptive pro-inflammatory responses (Figure 3B) [345,346]. Conversely, inhibition of EP2 signalling reverses cognitive aging and age-associated inflammation as well as the phagocytic defects in macrophages [346]. As EP2 receptors localized specifically to IBA1+ microglia in the hippocampal CA1 area and parietal cerebral cortex, PGE2-EP2 signalling inhibition could restore the phagocytic defects observed in LDAMs. Given that LDs are sites of PGE2 production, it is possible that the release of PGE2 from CNS LDs regulates myeloid glucose metabolism reprogramming and inflammation [346,347]. Further research is required to determine if LD accumulation in cells of the CNS is simply a result of inflammation or if LD biogenesis or catabolism can drive metabolic reprogramming in microglia to modulate cellular inflammatory responses as observed in macrophages of the peripheral nervous system.
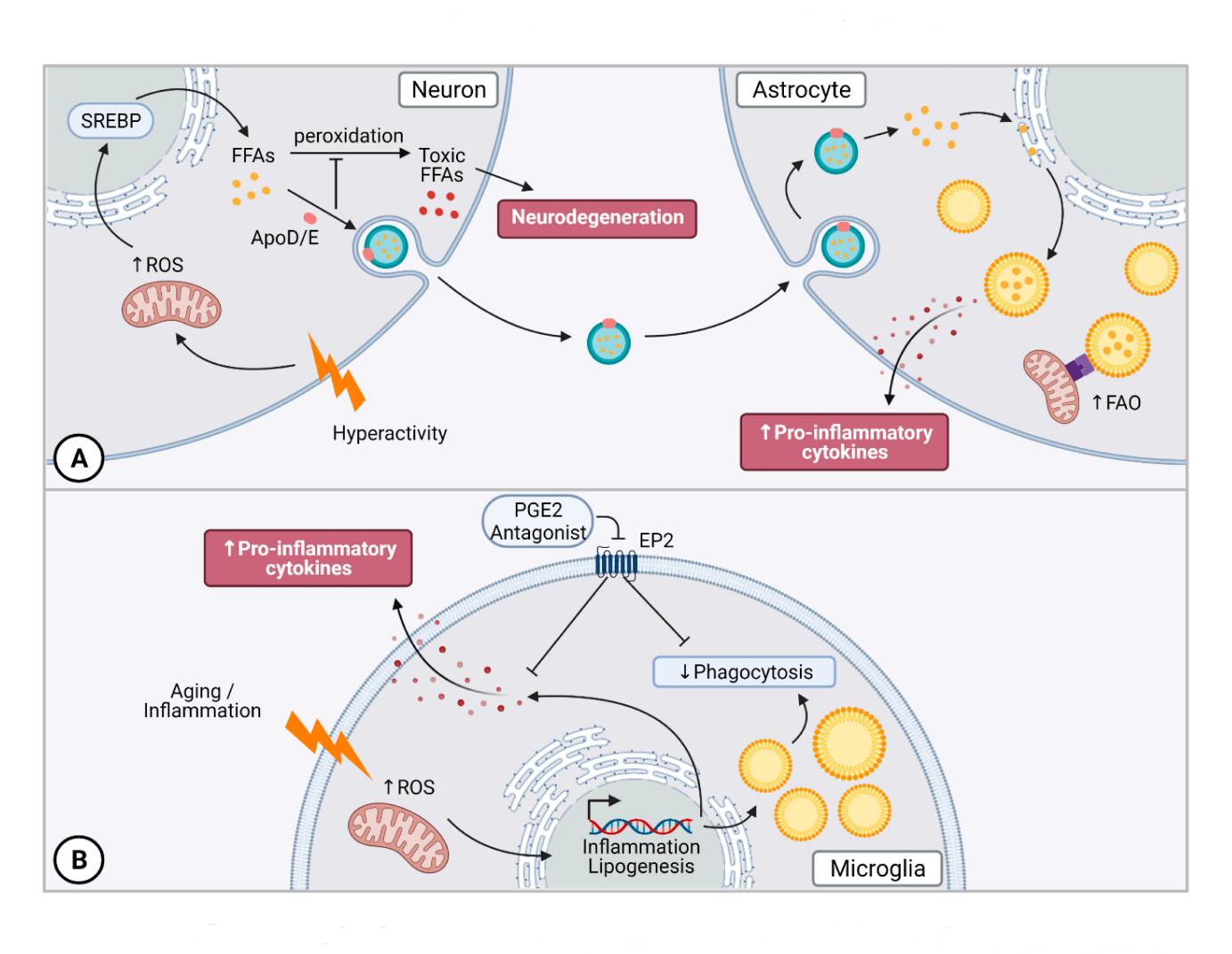 Figure 3. Lipid Droplets in the Central Nervous System. (A) Overstimulated neurons produce excess FFA through SREBP-mediated lipogenesis. Toxic FFA buildup leads to neurodegeneration that is prevented by shuttling excess lipids to glial cells via ApoD and ApoE to be integrated into LDs. Glial LD accumulation results in the release of pro-inflammatory cytokines. (B) LDAMs are a unique subset of microglia in the aging brain that accumulate LDs. They display defects in phagocytosis and increased pro-inflammatory cytokine secretion that can be reversed by inhibition of PGE2-EP2 signalling. Abbreviations: reactive oxygen species (ROS), free fatty acid (FFA).
Figure 3. Lipid Droplets in the Central Nervous System. (A) Overstimulated neurons produce excess FFA through SREBP-mediated lipogenesis. Toxic FFA buildup leads to neurodegeneration that is prevented by shuttling excess lipids to glial cells via ApoD and ApoE to be integrated into LDs. Glial LD accumulation results in the release of pro-inflammatory cytokines. (B) LDAMs are a unique subset of microglia in the aging brain that accumulate LDs. They display defects in phagocytosis and increased pro-inflammatory cytokine secretion that can be reversed by inhibition of PGE2-EP2 signalling. Abbreviations: reactive oxygen species (ROS), free fatty acid (FFA).
The past decades have witnessed a transformation in our view of cellular LDs, shifting away from simple passive lipid-storage organelles towards highly metabolic and interactive organelles with dynamic roles in cell metabolism and regulation of inflammatory responses. More than just bystanders resulting from excess lipid accumulation to prevent lipid-induced toxicity, LDs have emerged as signaling platforms that coordinate immune responses. The contribution of these previously underappreciated organelles to cellular signalling, antiviral responses, cell cycle regulation and antigen presentation will likely be the subject of exciting advances in the field of immunometabolism for the years to come. Delineating the regulation and function of LDs at a cellular level is of importance to understand the balance between protective and harmful LD accumulation in immune cells in the context of pathogenic, inflammatory, metabolic, cardiovascular, and neurodegenerative diseases. While much still remains unknown, it is clear that LDs play critical roles in cellular immunometabolism, regulating essential immune functions systemically in a disease- manner and providing promising therapeutic opportunities to develop novel, safe and effective approaches to fine tune LD cell biology to treat metabolic disorders.
The authors declare that they have no conflicts of interest.
This work was supported by funding from the Canadian Institutes for Health Research (PJT-391187 and Canada Research Chair to M.O.), the National Sciences and Engineering Research Council of Canada (Discovery Grant to M.O.), the University of Ottawa Cardiac Endowment Fund at the Heart Institute (Endowed Scholarship to D.B.), and a Queen Elizabeth II Graduate Scholarship in Science and Technology to V.V.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
117.
118.
119.
120.
121.
122.
123.
124.
125.
126.
127.
128.
129.
130.
131.
132.
133.
134.
135.
136.
137.
138.
139.
140.
141.
142.
143.
144.
145.
146.
147.
148.
149.
150.
151.
152.
153.
154.
155.
156.
157.
158.
159.
160.
161.
162.
163.
164.
165.
166.
167.
168.
169.
170.
171.
172.
173.
174.
175.
176.
177.
178.
179.
180.
181.
182.
183.
184.
185.
186.
187.
188.
189.
190.
191.
192.
193.
194.
195.
196.
197.
198.
199.
200.
201.
202.
203.
204.
205.
206.
207.
208.
209.
210.
211.
212.
213.
214.
215.
216.
217.
218.
219.
220.
221.
222.
223.
224.
225.
226.
227.
228.
229.
230.
231.
232.
233.
234.
235.
236.
237.
238.
239.
240.
241.
242.
243.
244.
245.
246.
247.
248.
249.
250.
251.
252.
253.
254.
255.
256.
257.
258.
259.
260.
261.
262.
263.
264.
265.
266.
267.
268.
269.
270.
271.
272.
273.
274.
275.
276.
277.
278.
279.
280.
281.
282.
283.
284.
285.
286.
287.
288.
289.
290.
291.
292.
293.
294.
295.
296.
297.
298.
299.
300.
301.
302.
303.
304.
305.
306.
307.
308.
309.
310.
311.
312.
313.
314.
315.
316.
317.
318.
319.
320.
321.
322.
323.
324.
325.
326.
327.
328.
329.
330.
331.
332.
333.
334.
335.
336.
337.
338.
339.
340.
341.
342.
343.
344.
345.
346.
347.
Boucher DM, Vijithakumar V, Ouimet M. Lipid Droplets as Regulators of Metabolism and Immunity. Immunometabolism. 2021;3(3):e210021. https://doi.org/10.20900/immunometab20210021

Copyright © 2021 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




