Location: Home >> Detail
Immunometabolism. 2020;2(4):e200032. https://doi.org/10.20900/immunometab20200032

1 Program in Molecular Medicine, University of Massachusetts Medical School, Worcester, MA 01605, USA
2 Laboratory of Adipose Tissue Biology, University of Mogi das Cruzes, Mogi das Cruzes, SP 08780-911, Brazil
3 Department of Biochemistry, Boston University School of Medicine, Boston, MA 02118, USA
* Correspondence: Miguel Luiz Batista Júnior; Tel.: +55-11-3091-7225; Fax: +55-11-3091-7402.
This article belongs to the Virtual Special Issue "Role of Local Adipose Tissue in Cancer"
Cancer-associated cachexia is defined by systemic inflammation, bodyweight loss, adipose tissue remodeling, and muscle wasting. Interestingly, until nowadays, the etiology for this syndrome still unclear. It is well known that multiple factors can contribute to adipose tissue remodeling, and longitudinal studies show that adipose tissue is affected early in the course of this syndrome. During cancer cachexia, adipose tissue remodeling is associated with adipocyte atrophy, impairment of fatty acid turnover, inflammation, reorganization of the extracellular matrix, and increased thermogenic gene programming of adipose tissue. Another attractive pathway is the adipose tissue lipolysis, which is the catabolic process that is leading to the breakdown of triglycerides stored in adipocytes and the release of fatty acids and glycerol. This pathway is highly involved in the adipose tissue wasting during cancer cachexia. Whole-body deletion of the genes that encode the lipolytic enzymes attenuates the effects of the syndrome on the reduction of body fat and muscle mass. These sets of changes, in addition to metabolites derived from this process, may be the initial trigger of the sequence of events that result in the remodeling and consequent dysfunction of adipose tissue during cancer cachexia. Therefore, this review aimed to investigate the main morpho-functional events that are resulting in adipose tissue remodeling in the context of cancer-associated cachexia.
Cancer-associated cachexia (CC) is common in the advanced stages of cancer and contributes to reducing the quality of life and life expectancy of patients [1]. CC is a significant cause of morbidity and mortality, affecting more than 80% of individuals with advanced cancer and accounting for more than 20% of deaths [2,3]. The severity of cachexia is not correlated with tumor size [4]. Although there are descriptions of cachectic individuals dating more than 2,000 years ago, this syndrome has been an increasing focus of attention in past decades [5]. Nevertheless, its etiology remains unknown, and no treatment can revert this condition [6,7]. This syndrome is characterized by reduced adipose tissue (AT) and skeletal muscle atrophy, accompanied by the establishment of a chronic inflammatory condition [8]. In regard to AT features, studies have shown that such tissue is affected by process of remodeling in response to various metabolic disorders [9]. In particular, more recently, some studies have shown that AT is changed early in the course of cachexia [10]. AT dysfunction is described as prevalent factors due to AT remodeling in response to cancer cachexia. In this scenario, the main modifications can be characterized such as; morpho-functional changes, chronic inflammation, alteration of white adipocyte phenotypes, and disruption of thermogenic profile.
Recently, an elegant recent review consistently described a consistent association between; skeletal muscle, adipose tissue, the bone, the liver, the neural system, and the gut with CC (3). This data set strongly suggests that CC is a multi-organ syndrome [11]. Among these organs, the communication between the adipose tissue and tumor is under intense investigation. Although the relationship between adipose tissue and tumor have been suspected for over 30 years [12,13], the detailed mechanisms have only recently been determined. In fact, AT is one of the largest organs in humans. Besides the energy storage function of AT, accumulating evidence demonstrates its unveiled roles within endocrinology, energy consumption, thermogenesis, stem cell pool, neuronal differentiation, and inflammatory regulation. However, the possible involvement of AT during cachexia development is largely overlooked.
Therefore, the present review aimed to describe state of the art related to the subject of interest by analyzing the central studies that have addressed the potential repercussions of AT remodeling during the development of cancer-related cachexia in both animal models and cancer patients.
Cachexia is understood as a complex metabolic syndrome associated with underlying diseases and is characterized by muscle atrophy and depletion of fat stores [14–16]. The main clinical symptoms are bodyweight loss in adults and impaired (substandard) growth in children (after exclusion of endocrine disorders) [8]. Anorexia, inflammation, insulin resistance and increased degradation of muscle proteins are frequently associated with cachexia [1]. However, the anorexia and weight loss of cachexia exhibit different characteristics compared with starvation, muscle mass loss due to aging (sarcopenia), primary depression, malnutrition and hyperthyroidism. The former is correlated with increased morbidity associated with asthenia and metabolic disorders [17,18].
Bodyweight loss is the foremost independent predictor of mortality in cancer patients [1,8], beginning with the loss of fat mass and lean body mass [3]. Over the last few years, the former has often has been proposed to precede and be affected more rapidly than the latter [16,19,20]. By the late 1980s and early 1990s, cachexia was being approached from a different perspective, leading to a new conception according to which it is considered a chronic inflammatory syndrome. It is currently believed that factors produced by both the tumor and host cause anorexia and the metabolic abnormalities that result in cachexia [17,21,22]. Nowadays, increased factors produced by both the tumor and host cause several metabolic abnormalities that have been described as an essential event that result in cachexia [1]. According to this consensus, the condition is categorized as pre-cachexia (early stage), cachexia or refractory cachexia (late stage), based on the severity of the following parameters: weight loss; the presence of metabolic disorders; anorexia and systemic inflammation.
A deeper understanding of the underlying mechanisms that underlie CC is vital for the development of new pharmacological and nutritional therapies. In this way, several studies [10,23–26] have suggested that AT is the target of local and systemic factors derived from the host and directly or indirectly produced by the tumor. These factors can be characterized, including; pro-cachectic factors (tumor necrosis factor α (TNF-α); interleukin (IL) 6, IL-1β; IL-8; interferon-γ (INF-γ); ciliary neurotrophic factor; leukemia inhibitory factor); anti-cachectic factors (soluble TNF-α receptor (sTNFR); soluble IL-6 receptor (sIL-6R); IL-1 receptor antagonist (IL-1RA); IL-4; IL-10; IL-15), and; tumor secreted factors (proteolysis-inducing factor (PIF); lipid-mobilizing factor (LMF); zinc-α2-glycoprotein (ZAG); toxohormone-L; leukemia inhibitory factor (LIF); anemia-inducing substance (AIS); parathyroid-hormone-related protein (PTHrP); heat shock protein 70 and 90 (HSP 70 and 90)), as such factors are involved in the etiology and progression of cachexia [11,20,27–29].
Adipose Tissue Inflammation as a Significant “Player”Considering the inflammatory model as the hypothesis to be tested, our group have recently demonstrated the significance of subcutaneous adipose tissue (scAT) as both an important source of inflammatory cytokine (especially, IL-6) and also a key source of adiponectin as a biomarker for cancer cachexia (clinical progression), as adiponectin exhibit a correlation with the magnitude of the total body mass reduction [23]. More recently, a consistent modification consisting of inflammatory cell presence and fibrosis in scAT induced by cancer cachexia was recognized in gastrointestinal cancer patients [30,31]. The scAT from these cancer patients was characterized by the presence of a crown-like structure composed of CD68 positive adipose tissue macrophages (ATMϕs) surrounding adipocytes and increased CD3 Ly, more evident in the fibrotic areas. Furthermore, some of these morphologic alterations were also present in the cancer patients (without cachexia), suggesting that adipose tissue inflammation may occur at an early stage of cachexia, even before the detection of pre-cachexia clinical features. Alterations in the adipose tissue inflammation seem to play a critical role in the fat mass reduction, in addition to other morpho-functional alterations related to the adipose tissue. Also, these changes appear to start quite early, long before any local (adipose tissue) and/or systemic (circulatory system) change can be detected.
However, although the description of the relevance of AT inflammation in clinical cachexia research is recent, this aspect has been characterized in the last decades, particularly in different experimental models of cachexia. In these models, AT remodeling, in particular, has been identified as a significant source of pro-inflammatory molecules, highlighting its importance not just as an energy stores depot, but as a metabolically active organ that may be relevant to the crosstalk between AT and other systems [3].
Nevertheless, most studies on this topic have been restricted to assessing inflammation from the systemic point of view, only investigating plasma parameters, while neglecting tissue inflammation and, particularly, the events that precede the appearance of these alterations. However they may significantly contribute to disorders that result in adipose tissue remodeling, such as impairment of lipid metabolism, tissue cell turnover and inflammation, fibrosis and subsequent systemic inflammation [23,31]. However, most studies related to the adipose tissue remodeling were restricted only to the assessment of inflammation in the systemic point of view (only investigating plasma parameters), while neglecting the local inflammation (adipose tissue) and, essentially, the events that precede the appearance of these alterations in the adipose tissue.
Additionally, considering the vital relationship among the infiltrated cells present in the tissue, the inflammatory mediators and the regulation of adipocyte metabolism of the adipose tissue, few or no studies have explored this relationship and the different stages of cancer cachexia to our knowledge.
In a general way, AT remodeling in CC comprise a set of morphological structural modification characterized by: (1) adipocyte atrophy, a result of a unbalance of lipids turnover, mainly due to increased lipolysis [15,32]; (2) modification of ECM component generally resulting in fibrosis [30,31]; (3) impairment of adipocyte cellular turnover (adipogenesis) [10,22]; (4) dysfunction of energy metabolism [33–35]; (5) enhanced inflammatory signaling [24,26,31,32,36]; (6) browning AT phenotype related to increase the thermogenic effect [20,37,38]. Consequently, this set of modifications results in a dysfunctional AT, capable of secreting a series of nutrients (especially of non-esterified fatty acids; NEFAs), adipokines [23,29,31], in addition to inflammation mediators that can be involved both in the beginning and in the establishment (amplification) of systemic inflammation.
Morphological AspectsAT, as a whole, is considerably heterogeneous ones. Its basic structure includes mature adipocytes, diverse immune cell populations, vascular cells, adipocyte precursor cells (APCs), a mostly uncharacterized stromal population, blood vessels, lymph nodes, and nerves [39,40]. Adipocytes are the primary cell type in AT. In contrast, the presence of other cell types varies as a function of the tissue location (mesenteric, epididymal, or subcutaneous), animal species, and disease (e.g., obesity and cachexia) [41]. Additionally, the importance of AT in the adiposity control, and its role of adipokines (leptin, adiponectin, and resistin, among others) are well established [9,42]. Still, alterations in the development and metabolism of adipocytes have been implicated in the pathogenesis of human immunodeficiency virus (HIV)-related lipodystrophy [43], On the other hand, few studies have addressed to understanding molecular mechanisms involved in the occurrence of lipodystrophy associated with other conditions, such as cancer cachexia. In the ongoing CC, the observed body weight loss predominantly results from a reduction in the fat and lean mass [15,44]. More recently, AT loss was found to precede the decline of the lean body mass, and thus, a more accurate understanding of this process began to emerge. Several alterations have been suggested as the cause of the reduction of AT mass, particularly in adipocytes atrophy. Those include; increased lipid mobilization due to increased lipolysis in adipocytes [14,15] reduction in lipogenesis, resulting from decreased LPL activity [21] and also impaired adipocyte turnover, most likely as a function of an adipogenesis-apoptosis imbalance in AT [45].
Thus, considering the factors that are most likely to be involved in fat mass loss, a recent study showed a time course of the main variables potentially involved in AT disorders from cachexia, emphasizing on the parameters related to adipogenesis, metabolism, and inflammation. The results showed that these alterations start early, at four days after the inoculation of Walker 256 tumor cells in Wistar rats [32,46,47] (Figure 1). This period precedes the appearance of the classic symptoms of cachexia, such as dyslipidemia and a reduction of the total body weight, as well as inflammatory alterations in the animals’ AT, which begin to predominate starting on day seven [10]. In Lung Lewis Carcinoma (LLC) tumor-bearing mice, up-regulation of genes related to lipid turnover and browning phenotype is evident even before the detection of the body weight loss of the animals [20,37,38].
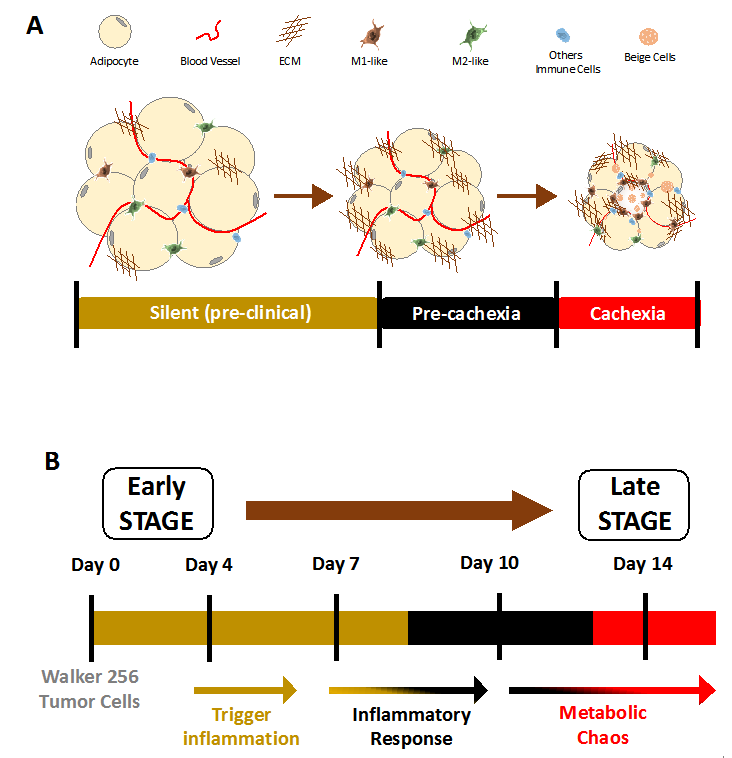 Figure 1. Model of cachexia development from a translation point of view. (A) Morpho functional changes in adipose tissue described in cancer cachexia patients. These alterations are associated with the stages of syndrome development. (B) Compilation of main metabolic and inflammatory changes described in an experimental model of cachexia induced by Walker 256 tumor. Adapted from [47], an open access article distributed under the Creative Commons Attribution 3.0 International License (http://creativecommons.org/licenses/by/3.0/).
Figure 1. Model of cachexia development from a translation point of view. (A) Morpho functional changes in adipose tissue described in cancer cachexia patients. These alterations are associated with the stages of syndrome development. (B) Compilation of main metabolic and inflammatory changes described in an experimental model of cachexia induced by Walker 256 tumor. Adapted from [47], an open access article distributed under the Creative Commons Attribution 3.0 International License (http://creativecommons.org/licenses/by/3.0/).
Similarly, using several animal models of cachexia (syngeneic and genetic), K5-SOS-F transgenic mice showed a reduction in fat mass and spleen enlargement in the pre-cachexia, that is, before the detection of body weight loss and the development of cachexia [37]. Thus, the temporal characterization of some of the alterations occurs in different AT depots in the course of cachexia. It has effectively pointed to specific pathways and mediators that might be involved in the earliest changes, in addition to serving as biomarkers for the clinical progression of cachexia.
As mentioned above, the accentuated reduction of fat mass is a significant clinical of cancer-related cachexia. Although, there is no information whether these alterations depend on the location of the AT involved (e.g., visceral or subcutaneous). In this aspect, a recent study using the same animal model of cachexia (Walker 256 tumors cells-induced) showed that visceral fat depots are affected in different ways. Thus, epididymal AT exhibited higher relative weight, while retroperitoneal AT did not show any change [48]. Furthermore, in cachectic patients, assessment of the AT area employing computed tomography in humans showed that visceral AT (omentum) showed to be reduced in cachectic individuals with gastrointestinal carcinosarcoma compared with controls [49]. On the other hand, in individuals with gastrointestinal cancer, scAT seems to be more affected than visceral AT (omental and mesenteric), at least considering tissue inflammation parameters [50].
Extracellular Matrix RearrangementExtracellular matrix (ECM) remodeling is the result of the processes of matrix synthesis and degradation. It occurs under physiological (e.g., tissue repair) and pathological conditions (e.g., inflammation). The ECM is mainly composed of a complex network of multifunctional and structural molecules, including various collagen isoforms, adhesive glycoproteins, and proteoglycans [51]. This network provides support to cells that control their migration, proliferation and differentiation. Furthermore, ECM can serve as a reservoir of cytokines and other growth factors, which are secreted depending on the pathological condition [47]. ECM remodeling plays a central role during adipogenesis. Although the molecular mechanisms are partially comprehended, ECM remodeling occurs parallel with the activation and/or repression of a transcriptional “network” involved in adipogenesis, which may be activated or repressed due to extracellular stimuli [52]. The adipogenesis is associated with a reduction of the fibronectin-rich matrix and basal lamina formation in the 3T3-L1 mouse cell line [53,54]. This condition is an example of the relationship between structural changes in ECM and adipocyte differentiation.
Regarding the different cell types present in AT, progenitors cells and preadipocytes might participate in interstitial fibrosis development, which mostly involves type I collagen and fibronectin, as observed in the scAT of obese individuals. As a result, preadipocytes might significantly contribute to the pathological alterations in the AT of such individuals. Besides, the increase in the number of infiltrated macrophages suggests a direct relationship with the changes in the ECM (collagen deposition) and the metabolic alterations (insulin resistance) that take place in the AT of obese individuals [55]. In CC, collagen content deposition, an increase in the number of infiltrated cells and insulin resistance have also been observed. However, very little is known regarding the possible relationship between these cell types (e.g., fibroblasts, myofibroblasts, preadipocytes, immune cells, and others) and the processes that lead to ECM remodeling.
In the cachexia experimental model, shrunken adipocytes and increased collagen-fibril content in AT were reported in MAC16 tumor bearing-mice [22]. Our group recently showed that the total type I collagen content of the scAT is rearranged in cachectic individuals with gastrointestinal cancer, which is associated with an increase in macrophage and lymphocyte infiltration. Interestingly, the total collagen content exhibited discrete changes in cancer patients without cachexia (weight‐stable), but the expression of the CCL2 (MCP-1) gene was upregulated [31]. Other interesting findings demonstrate that ECM remodeling of AT in cancer cachexia results not only in augmented collagen fiber content, but also followed by excessive synthesis of mature elastic fibers, besides enhancement of collagen type I and III in the AT from cachectic patients [30,31]. Also, the presence of fibrosis was also associated with an increased number of myofibroblasts and an activated TGFβ/SMAD pathway in the subcutaneous AT of gastrointestinal cancer cachectic patients [30]. These findings show a morphological rearrangement that leads to AT remodeling in cachectic patients, although slightly, before the onset of the earliest characteristic symptoms of cachexia (Figure 2). However, to the best of our knowledge, no study has yet investigated the causes and repercussions of AT remodeling in the course of cachexia in full detail [47].
Metabolic DysfunctionIt is thought that the loss of body weight during cancer cachexia is the result of profound changes in the metabolic pathways of tissues and organs, which cannot be explained by increased energy expenditure or malnutrition [3]. In cachectic patients, loss of fat mass precedes muscle wasting and is invariably more pronounced, or even evident in the absence of lean tissue loss [56]. In this sense, the rapid dysfunction of the adipose tissue has been gaining great importance in the beginning and progress of many changes induced by the syndrome.
As described above, different mechanisms have been proposed to explain changes in AT during cachexia, such as, increased lipolytic activity and decreased activity of lipoprotein lipase (LPL) [11,32,56,57]. Regarding the increase in lipolysis, lipolytic factors or hormones, such as tumor necrotic factor α (TNFα) [58], interleukin-6 (IL-6) [59], Zinc-α2-glycoprotein (ZAG) [27], catecholamines, and natriuretic peptides [14], could explain the enhancement of lipolysis during cancer cachexia. More recently, a study shows that leukemia inhibitory factor (LIF), a C-26 tumor cells-secreted molecule, mediates lipolysis in an ATGL-dependent pathway [29].
AT mass is determined by lipid turnover, i.e., the balance between incorporation and removal of TG into adipocytes. Lipolysis is the most critical factor for lipid removal [45]. During cancer cachexia, human and animal models of cancer cachexia have demonstrated an increased rate of lipid mobilization [16]. Longitudinal studies have shown that cachectic patients start to lose adipose tissue mass before wasting of the muscle mass can be detected [19]. In addition, an earlier rate of adipose tissue loss is believed to be associated with shorter survival time during cancer progression [60].
Regarding the mechanisms that may be involved in the reduction of fat mass during cachexia, the increase in lipolysis seems to be the most evident and is being described as an increasing frequency [14,16]. Das and colleagues found that ATGL and HSL knockout mice exhibit resistance to the development of cancer-associated cachexia (16). While HSL contributes, ATGL-mediated lipolysis of triglycerides appears to be most relevant. Further confirmation for ATGL leading role was provided by studies in ATGL and HSL deficient mice bearing the cachexia inducing B16 melanoma and LLC tumor cells. In particular, the attenuation of fat mass loss showed a positive correlation with the muscle mass maintenance. An interesting positive correlation between ATGL activity and the severity of cachexia was also observed. This study corroborates findings previously reported in cachectic patients [19], thus increasing the evidence that demonstrates the role of AT as a possible therapeutic target during the cancer-cachexia syndrome. More recently, in mice syngeneic to the C26c20 tumor line, it was identified LIF as a factor secreted from cancer cells that induce lipolysis. In this animal model, LIF can induce adipocyte ATGL-mediated lipolysis through GP130 and JAK/STAT activation. Interestingly, LIF showed an effect independent of changes in food intake and leptin levels [61].
To illustrate that, a study evaluated the lipolytic capacity of isolated adipocytes in two different periods (4 and 14 days) after tumor cell inoculation. Mesenteric and retroperitoneal fat were analyzed based on previous studies that demonstrated their relevance during cancer cachexia syndrome. Day 4 was selected to perform the analysis, also based on previous results [10], it was observed that in this specific period there was a profound negative regulation of the genes involved in the metabolism of adipocytes. In addition, no changes were found in the morphological and inflammatory parameters evaluated. Interestingly, the cachectic mice demonstrated a reduction of basal lipolysis four days after the tumor cell inoculation, while on day 14, the animals exhibited an increase in the basal lipolysis rate (Figure 1). These findings support the results of other studies showing increased lipolysis in the subcutaneous adipose tissue of cachectic patients [14,62].
In this sense, another relevant aspect was that the deregulation of lipolysis (in vitro) revealed a different profile, depending on the disease progression. In an early stage, the basal rate of lipolysis was reduced and an increase in p-HSL (Ser565) was observed, which is regulated by the activation of AMPK and, consequently, inhibits HSL activity. Cachectic patients show a decrease in basal lipolysis with elevated ex vivo responses to catecholamine and natriuretic peptides [44,63]. Another study showed that the lack of AMPK activity as a common feature of adipose tissue dysfunction during cancer cachexia in mice triggered by the induction of CIDEA and the consequent degradation of AMPK in this tissue [33]. The authors suggest a possible cachexia treatment, using a peptide specifically targeting the adipose tissue AMPK-CIDEA interaction to prevent adipose tissue wasting during cancer cachexia syndrome.
Adipogenesis ImpairmentMature adipocytes can be characterized as a post-mitotic cell [64]. Annually, ~8% of adipocytes are turned over to match the rates of cell death [65]. Adipogenesis may be defined as the process of the differentiation of progenitors adipocytes cells (APCs), mainly preadipocytes, into new adipose cells that are able to store TG and synthesize and secrete hormones and adipokines (mature adipocytes) [66]. In fact, the development of obesity-related metabolic disorders, such as peripheral insulin resistance, hyperlipidemia and type 2 diabetes is connected to impaired adipogenesis [67]. The process of adipose cell differentiation involves the activation of a cascade of transcription factors that coordinate the expression of genes responsible for adipocyte function [67,68]. Local and endocrine factors might regulate adipogenesis by modulating these transcriptional events [47,68].
In a simplified way, the AT cellular turnover is regulated by a balance between the growth/differentiation (adipogenesis) and the death of its cells (generally by apoptosis) [35,65]. In the course of cachexia, the impairment of adipogenesis was recently addressed. In this aspect, studies have elucidated the adipogenic marker profile during the development of cachexia syndrome [27]. It has been known that adipogenic genes are downregulated in CC from epididymal AT [10,22] and retroperitoneal AT [10]. Recently, the possible involvement of cellular turnover in the cachexia process was addressed.
Retroperitoneal AT showed reduced Pref1/Adiponectin ratio of gene expression from the stromal vascular fraction and increased pro-apoptotic CASPASE3 and CASPASE9 protein levels from the same tissue (35). On the other hand, subcutaneous AT apoptosis markers did not change in cancer patients (49). In this scenario, despite consistent evidence about the impairment of cellular turnover, especially adipogenesis, few studies have investigated this aspect in more detail.
Using a co-culture system to mimic the effects of CC on adipocytes, a recent study has analyzed in vitro adipogenesis in 3T3-L1 cells [69]. LLC cells secreted factor promoted a decreased volume of the lipid droplets, compromising its maturation process (adipogenesis). This result was followed by the downregulation of adipogenic and lipolytic gene expression, increased in apoptosis markers and proinflammatory cytokines secretion from both tumor cells and adipocytes. These data suggest that the presence of the tumor cells was able to inhibit adipocytes maturation with was associated with the increased levels of inflammatory cytokines. These same results were observed in primary adipocytes, cultivated with conditioned medium from LLC cells.
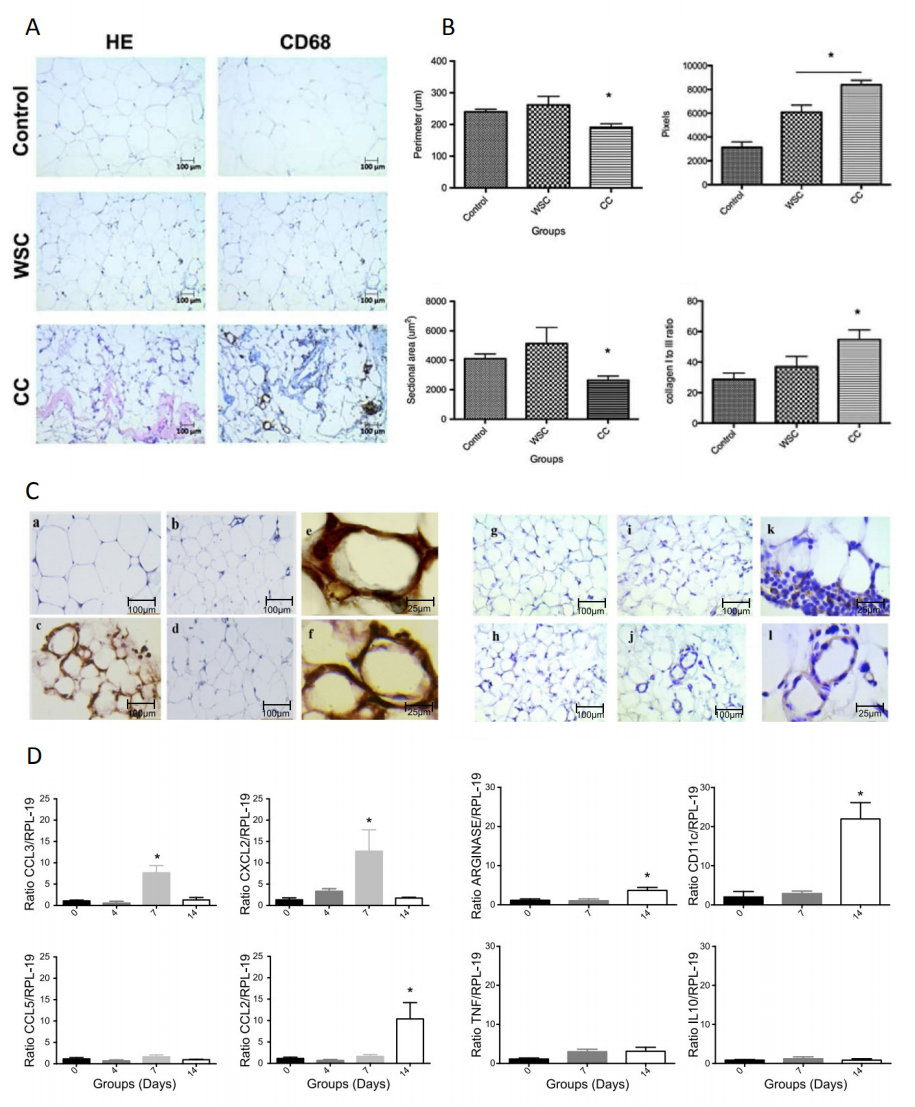 Figure 2. (A) Identification of different immune cell types present in subcutaneous adipose tissue obtained from cancer cachexia patients. Serial sections of weight‐stable subjects and control, weight‐stable cancer (WSC) patients and cancer cachexia (CC) patients were stained with markers of macrophages (CD68). (B) Morphometric analysis of the sectional area, the perimeter of adipocytes and total collagen quantification from different experimental groups. (C) Immunohistochemistry of mesenteric adipose tissue sections stained with CD11b (left panels: (a) control; (b) day 4; (c) day 7; (d) day 14; (e–f) day 7) and CD68 (right panels: (g) control; (h) day 4; (i) day 7; (j–l) day 14). (D) Quantitative PCR of mRNA gene expression of cell infiltration markers (CCL2, CCL3, CCL5, and CXCL2) from SFV of mesenteric adipose tissue. Values are mean ± SEM. * P < 0.05 vs control subjects. Figure 2A,B adapted from [31], an open access article distributed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/). Figure 2C,D adapted from [32]. copyright © 2017 John Wiley & Sons, Inc.
Figure 2. (A) Identification of different immune cell types present in subcutaneous adipose tissue obtained from cancer cachexia patients. Serial sections of weight‐stable subjects and control, weight‐stable cancer (WSC) patients and cancer cachexia (CC) patients were stained with markers of macrophages (CD68). (B) Morphometric analysis of the sectional area, the perimeter of adipocytes and total collagen quantification from different experimental groups. (C) Immunohistochemistry of mesenteric adipose tissue sections stained with CD11b (left panels: (a) control; (b) day 4; (c) day 7; (d) day 14; (e–f) day 7) and CD68 (right panels: (g) control; (h) day 4; (i) day 7; (j–l) day 14). (D) Quantitative PCR of mRNA gene expression of cell infiltration markers (CCL2, CCL3, CCL5, and CXCL2) from SFV of mesenteric adipose tissue. Values are mean ± SEM. * P < 0.05 vs control subjects. Figure 2A,B adapted from [31], an open access article distributed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/). Figure 2C,D adapted from [32]. copyright © 2017 John Wiley & Sons, Inc.
In general, despite the results obtained in the experimental model had demonstrated cachexia-induced impairment of adipogenesis in vitro, further analysis using tracer lineage animal model would be a useful approach for a more detailed investigation. Besides, failure in adipogenesis may precede the appearance of the classic signs of cachexia as well as the firsts signs of AT inflammation (mentioned above), such as increases in infiltrated macrophages and the production of inflammatory cytokines [24] (Figure 2). Thus, the factors that “silence” the genes involved in the differentiation of preadipocytes and maintenance of adipose cell turnover in AT might play a central role in the genesis of the damaging changes that occur in the adipose tissue of individuals with cachexia [47].
Role of InflammationDuring the development of the CC occurs a metabolic and morpho-functional dysfunction that results in AT remodeling (Figure 2). Both animal models and cancer cachexia patients consistently show this feature. Besides, another aspect to be highlighted of cachexia-induced AT remodeling is the establishment of AT inflammation that is characterized by increased recruitment of ATMϕs [38]. Such an inflammatory profile seems to be more evident in the end-stage of cachexia, as most of the characteristic changes of this syndrome are already established at this time [10,46,70].
Thus, using the cachexia model induced by Walker 256 cells as a prototype to characterize the main signs and symptoms involved in the development of cachexia (Figures 1 and 2), increase in M1-polarized macrophages can be observed as the main subset of cell infiltrates in visceral AT [32]. Also, the upregulation in the CCL3 and CXCL2 gene expression at seven days after tumor cell injection, suggests the presence of neutrophils in the various AT depots evaluated. The presence of CD11b-positive cells, to detect the presence of neutrophils, was observed at the same time. Thus, it does characterize that inflammation starts early (on day seven) and is established by day 14. In this model, cachexia becomes evident (e.g., a reduction of the total body and fat mass, dyslipidemia, hypoglycemia) between day 13–15 after tumor cell injection. In another experimental model, these findings were presented in greater detail in cachexia induced by LLC cells, showing ATMϕs polarization tends to be directed to M1 phenotype [38].
In the last few years, the importance of the relationship between inflammation and CC, and systemic inflammation, in particular, has drawn attention. However, there is also no consensus concerning the role of AT inflammation in the establishment and development of cancer cachexia, among cancer patients in particular [14,49]. Limitations in experimental designs, the selection of control groups, and the techniques used to analyze inflammation markers have most likely been responsible for preventing a more precise investigation of the presence of inflammation in AT [47]. Addressing this question, in cachectic patients with gastrointestinal cancer, upregulation of gene expression of phenotypic markers of ATMϕs and inflammatory cytokines, such as IL-6 and TNF-α [23]. Interestingly, higher gene levels of IL-6 were positively correlated to higher plasmatic levels of this same cytokine in cancer cachexia patients, indicating that scAT may be an important source of inflammatory mediators. More recently, the same group showed an increase of ATMϕs forming crown-like structures in the same AT depot from cachectic patients [31] (Figure 2), which is a characteristic finding in fat tissue in experimental models of obesity and obese patients [71]. Also, increases of chemoattractant gene expression for ATMϕs in scAT, such as CCL2 was detected only in weight stable cancer patients (without cachexia), showing no changes in cachectic ones. However, despite the relevance of local inflammation, in AT, in particular, the mechanisms responsible for both cachexia and inflammation remain to be elucidated [47].
Currently, the presence of inflammatory infiltrates in different fat depots phenotype is well established in clinical research and the experimental model of cancer cachexia. On the other hand, for the most part, these analysis studies evaluate the whole AT. They do not approach tissue in a usual manner, especially considering the heterogeneity of the different cell types of the fraction of the vascular stroma (single-cell level). Moreover, the presence of inflammation seems to precede the ECM proteins' rearrangement establishment, particularly the collagen content. In this aspect, the turnover lipids disorder appears to precede all previous events, placing the adipocyte dysfunction ability to store/secrete fatty acids as one of the first events (Figure 1). Thus, specific characterization of factors secreted by the tumor and host and the possible interaction with wasting tissues are significant challenges for understanding the inducing molecules and pathways related to the AT remodeling.
Thermoregulatory Response (Possible Role of AT Browning)Regarding the main changes that result in the AT remodeling, it has recently been demonstrated that cachexia induces AT browning [20,37,38]. In relation to that, chronic inflammation and β-adrenergic activation are involved in the cancer cachexia pathogenesis [20,37,72,73]. AT browning has also been described an increase in total caloric expenditure [74] and a potential therapeutic approach reducing of body fat [20,37]. In this sense, during cancer cachexia, the browning phenotype’s presence has shown to be detected very early [20] in different experimental models [20,37,38], demonstrated the by up-regulation of gene and protein levels of UCP1. Some studies have also demonstrated an intense alteration in the adipose tissue metabolism, since this process is related to the increase in energy expenditure, weight loss and mobilization of fatty acids by adipocytes [20,37].
In relation to thermogenesis, body temperature is hugely reduced when the main clinical signs are already established (refractory cachexia). Interestingly, this hypothermia picture has previously been described in the Walker 256 tumor-induced cachexia model, in the final stages of cachexia [75]. Besides, despite most of the experimental studies (animal model) showed an increase in energy expenditure (EE) in response to induced cachexia [20,37], Rolhm et al. (33) recently showed that there is no change in EE in LLC-bearing mice. This is due to the differences between; type of tumor, the number of cells inoculated, and energy consumption analysis models. In this sense, additional studies should be addressed to clarify the main metabolic changes resulting and or, in some way, are connected to the thermogenic adjustments induced by cancer cachexia. Despite the browning phenotype being described in samples of visceral fat from cachectic patients [37], analysis in a larger cohort and more studies with human data are required to understate the possible physiological repercussions during cancer cachexia.
As mentioned above, although several well-designed studies demonstrate AT browning induced by CC, little is known about the inducing factors and cellular mechanisms responsible for this process. In this sense, Kir et al. [20] showed that tumors, or factors secreted by them, can directly activate thermogenesis in beige cells through the secretion of the thyroid hormone-related protein (PTHrP). This molecule has been identified in culture supernatants from a specific (clonal) selection of LLC cells from mice. It has been shown to be able to induce Ucp1 expression categorically. In addition to the direct activation of AT browning by tumor-derived PTHrP, systemic inflammation and activation of the β-adrenergic pathway represent complementary mechanisms involved in inducing the browning phenotype in AT in response to CC [37]. Also, more recently, some molecules, such as BMP7 (Bone Morphogenetic Protein 7), Pten (phosphatase and tensin homolog), and FGF21 (Fibroblast growth factor 21) have been shown to induce beige cells [76]. On the other hand, few studies have related the possible role of these molecules to CC's browning.
Regarding browning phenotype and the consequent increase in thermogenesis in AT, most of the studies have shown it is dependent on the enhance in UCP1. However, recently, an elegant study carried out in an experimental model of cachexia induced by different tumor cells [33], chemical denervation in BAT and knockout model for UCP1, demonstrated that the increase in thermogenesis in AT occurs via an independent pathway from UCP1 and that other metabolic events, such as increased lipid turnover, would have a relevant role in the set of changes that result in metabolic chaos and, consequently, in the reduction of body mass in cachexia.
Additionally, considering the relevance of the AT browning process as a relevant event in the different stages of cachexia, some pharmacological interventions have shown. Treatment with β- adrenergic antagonists, non-steroidal anti-inflammatory drugs (Sunlidac), and more recently, treatment with TLR4 antagonist (Simvastatin), are efficient in attenuating AT browning in CC, in addition to preserving the body mass in tumor-bearing animal [37]. The latter showed more significant effects, and the only one that showed increased survival.
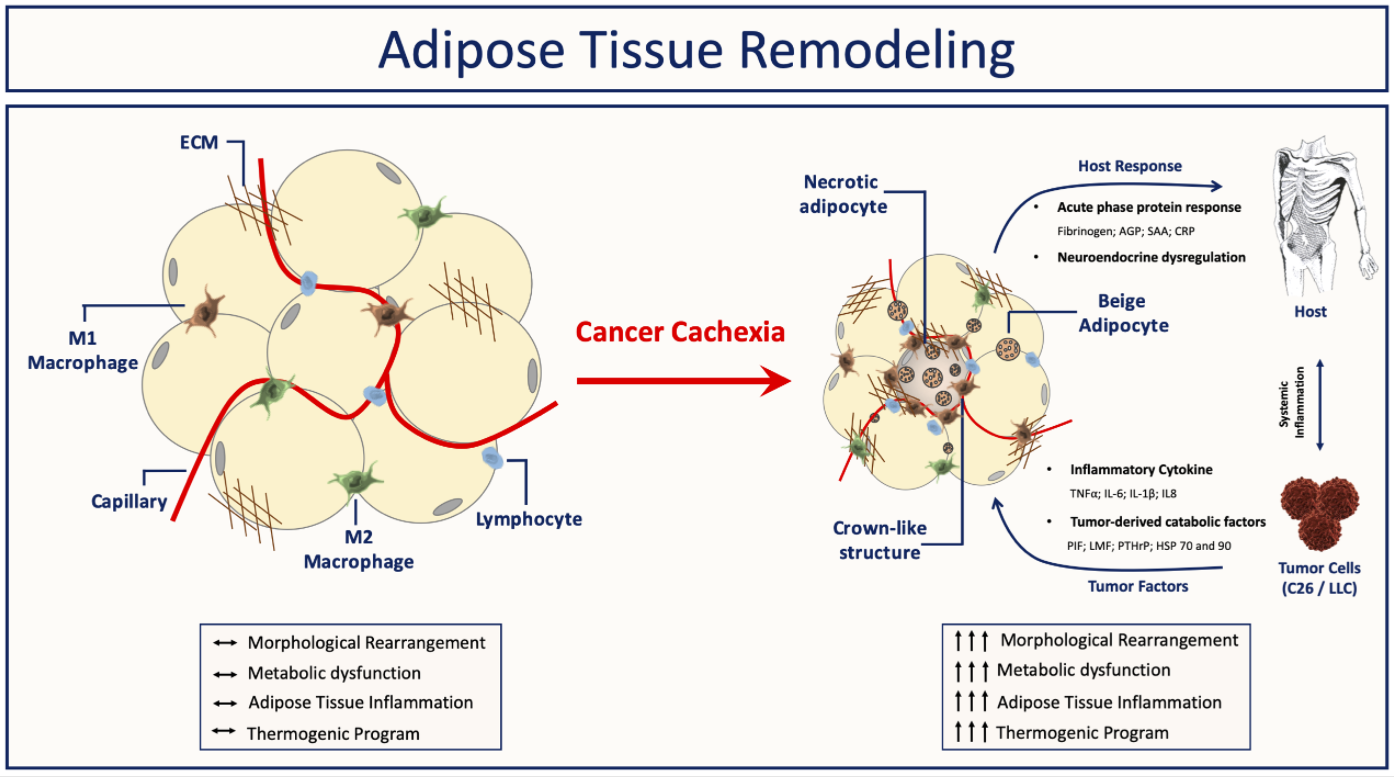 Figure 3. Summary and proposed model: Factors produced by both the tumor and host are essential to cause the metabolic abnormalities that result in cachexia. AT is affected very early, and with the ongoing of its dysfunction, in parallel to syndrome development, it results in AT remodeling. Adipose tissue inability to accurately sense and respond to inflammatory stimuli leads to adipose tissue atrophy, increased fibrosis, immune cell infiltration, and an increase of beige cells.
Figure 3. Summary and proposed model: Factors produced by both the tumor and host are essential to cause the metabolic abnormalities that result in cachexia. AT is affected very early, and with the ongoing of its dysfunction, in parallel to syndrome development, it results in AT remodeling. Adipose tissue inability to accurately sense and respond to inflammatory stimuli leads to adipose tissue atrophy, increased fibrosis, immune cell infiltration, and an increase of beige cells.
In summary, AT is significantly affected during cancer cachexia in both different experimental models and clinical studies (Figure 3). In concern morpho-functional disorders, early adipocyte lipid turnover dysfunction of AT, and increases in immune system cells, followed by increased local production of inflammatory mediators, and remodeling of ECM components have been described as the significant changes that result in AT dysfunction. In addition to the modifications above, some studies have shown that cachexia induces browning of AT, although its function is still not well characterized. Nevertheless, different studies have consistently shown that the modifications in the adipocyte metabolism begin quite early, and the metabolites derived from this process may be the initial (sterile) trigger of the sequence of events that results in the AT remodeling and its consequent dysfunction in cachexia. Finally, understanding the initial stimulus that triggers AT dysfunction is required, in particular, morpho-functional and inflammation once evidences also indicating to have a significant role in this progression of cachexia syndrome, as well as be a potential modulator of the process that could be explored therapeutically.
FH and MLBJ, contributed equally to the discussion of content, researching data for the article, writing the article and reviewing and editing the manuscript before submission.
The authors declare no conflicts of interest.
This work was supported by São Paulo Research Foundation (FAPESP) Grants: 2015/19259-0, 2017/24615-5, 2018/20905-1 and CNPq 311319/2018-1 to MLBJ. This article’s contents are solely the responsibility of the authors and do not necessarily represent the official views of FAPESP and CNPq. The authors gratefully acknowledge the commitment and support by the American Diabetes Association (ADA) Grant #1-19-PMF-035 to F.H.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
Henriques F, Batista Júnior ML. Adipose Tissue Remodeling During Cancer-Associated Cachexia: Translational Features from Adipose Tissue Dysfunction. Immunometabolism. 2020;2(4):e200032. https://doi.org/10.20900/immunometab20200032

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




