Location: Home >> Detail
Immunometabolism. 2020;2(3):e200022. https://doi.org/10.20900/immunometab20200022
 ,
Theresa Ramalho 3
,
Theresa Ramalho 3
1 Department of Intensive Care Unit, Renmin Hospital of Three Gorges University, The First People’s Hospital of Yichang, Yichang 443000, Hubei, China
2 Amsterdam UMC, Vrije Universiteit Amsterdam, Department of Molecular Cell Biology and Immunology, Amsterdam Cardiovascular Sciences, Cancer Center Amsterdam, Amsterdam, 1081HZ, The Netherlands
3 Department of Immunology, Institute of Biomedical Sciences, University of Sao Paulo, Sao Paulo 05508-900, Brazil
* Correspondence: Jan Van den Bossche.
The increased incidence of cancer has been attributed to raised numbers of individuals with obesity/overweight worldwide. Different types of cancers in obese individuals have poor prognosis, high remission rate and resistance to traditional therapies. Literature has identified that lipid metabolism in lipid-laden immune cells, including macrophages, abrogates anti-tumoral responses. In this review we highlight the main metabolic features observed in macrophages in obesity and cancer, and discuss how metabolic changes during metabolic diseases elicits a phenotype in macrophages that could promote malignant progression. This perspective could support new diagnosis and treatment strategies for cancer patients in immunometabolic predisposed condition.
Macrophages are a central component of innate immunity. These cells have different origins and exhibit great functional diversity [1]. They are found in all tissues and play vital role in development, maintenance of homeostasis, in pathologies such as infections, metabolic diseases, cancer, and in resolutive responses [2,3]. For many years, macrophage phenotypes have been categorised into different subtypes. In this sense, macrophages are often broadly subdivided into classic or inflammatory M1 macrophages activated by TNF-α, LPS and/or IFN-γ, with microbicidal and tumoricidal activity; while regulatory or alternatively activated M2 macrophages induced by IL-4, IL-13, IL-10 and/or TGF-β, are involved in parasite containment, promotion of tissue remodeling, tumor progression and immunoregulatory functions [4]. Macrophages show metabolic reprogramming in response to activation, and in this simplified view, M1 subsets favor glycolysis, pentose phosphate pathway, and fatty acid synthesis, whilst M2 types are more dependent on an intact Krebs cycle and a functional oxidative phosphorylation [5]. The distinct metabolic features of macrophage subsets not only regulate their polarization and function, but also determines their plasticity and repolarization capacity [6]. In vivo, different environmental signals promote a spectrum of activation states that extends the M1/M2 model [7,8]. Different states of activation in macrophages observed in metabolic diseases may favor tumor growth. In this scenario, the nutritional condition of an individual can affect the composition of substrates that feed and functionally alter macrophages within different microenvironments, including tumor-associated macrophages (TAM) in the tumor microenvironment (TME). For example, altered lipid metabolism observed in individuals with metabolic diseases lead to accumulation of lipids in circulation and tissues, which further influences the activation state of macrophage populations and promote phenotypes that could favor tumor progression [3,9]. This may explain part of the reason why metabolic diseases that have hyperlipidemia as a central feature, promote cancer development followed by a poor prognosis [3]. The purpose of this review is to discuss how intracellular metabolic changes in macrophages during metabolic disease lead to phenotypes that could promote tumor development and progression. The prevalence of obesity is increasing worldwide, therefore mechanistic comprehension of the obesity-cancer relationship is urgently needed to provide insights for new diagnosis and treatment strategies in obesity-associated cancer patients.
The World Health Organization (WHO) has reported obesity as one of the most prevalent global health issues with 35% of adults around the world being overweight or obese [10]. According to the World Obesity Federation, more than 70% of obese cases are associated with other metabolic syndromes, such as atherosclerosis and type 2 diabetes [11]. In obesity, the hypertrophic adipose tissue is characterized by the infiltration of immune cells that contribute to a chronic state of both local and systemic inflammation directly linked to insulin resistance [12]. Among immune cells, macrophages are the most predominant cells in hypertrophic adipose tissue. These cells can originate from recruited monocytes and local hematopoietic stem/progenitor cell populations in mouse white adipose tissue [13,14]. Adipose tissue macrophages (ATM) from different origins belong to different subtissular niches described below (Figure 1).
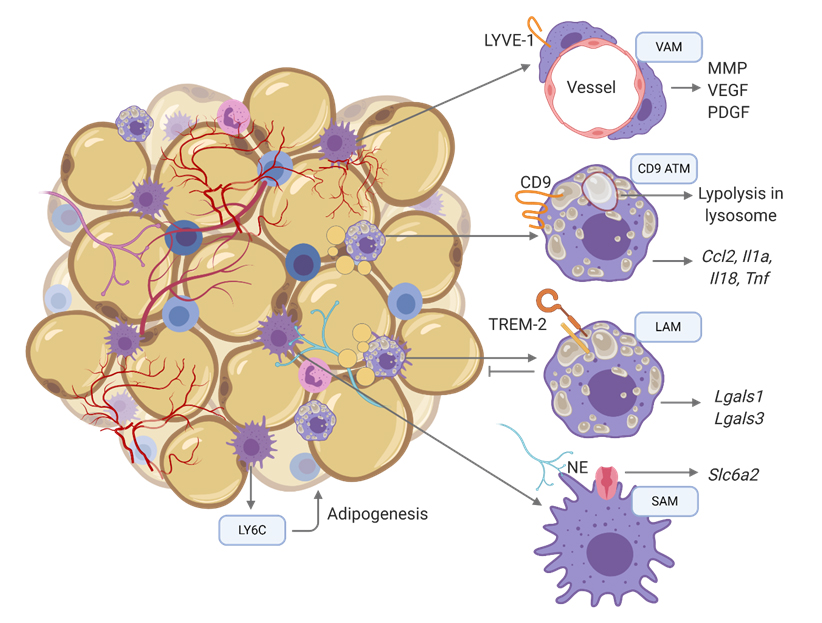 Figure 1. Distinct macrophage populations in obese adipose tissue. Obese fat tissue of mice and humans contain multiple distinct populations of ATMs with unique tissue distributions, transcriptomes, and functions. ATM, adipose tissue macrophage; VAM, vascular associated macrophage; LAM, lipid associated macrophage; SAM, sympathetic associated macrophage.
Figure 1. Distinct macrophage populations in obese adipose tissue. Obese fat tissue of mice and humans contain multiple distinct populations of ATMs with unique tissue distributions, transcriptomes, and functions. ATM, adipose tissue macrophage; VAM, vascular associated macrophage; LAM, lipid associated macrophage; SAM, sympathetic associated macrophage.
Subpopulations of ATMs can be located in distinct regions of the adipose tissue exhibiting distinct transcriptional signatures specific to their location (Figure 1). In hypertrophic adipose tissue, macrophages engulf dying adipocytes, configurating crown-like structures (CLS) composed by lipid-loaded macrophages [15]. In this population, CD9+ CLS macrophages express genes related to lysosomal-dependent lipid metabolism, and have a pro-inflammatory phenotype [16]. Another subpopulation of CLS is the lipid-associated macrophages (LAM) derived from circulating monocytes, that express an immune suppressive phenotype and prevents adipocyte hypertrophy through a TREM-2 (Triggering Receptor Expressed on Myeloid Cells 2)-mediated mechanism [17]. Yet the sympathetic neuron-associated macrophages (SAM) scavenge excessive norepinephrine (NE) produced by sympathetic neurons of the obese adipose tissue [18]. Monocyte-derived Ly6C-expressing macrophages (CD11b+Ly6c+) uniformly distributed throughout the adipose interstitium promote adipogenesis and tissue reorganization [16]. On the other hand, resident or monocyte-derived vasculature-associated macrophage (VAM) express lymphatic vessel endothelial receptor 1 (LYVE1) and secrete tissue remodeling factors and factors promoting the formation of endothelial cell tubes [19], similar to conventional TAM. In summary, CD9 and SAMs ATMs can be considered pro-inflammatory, while VAMs, LAMs and CD11b+Ly6c+ have an anti-inflammatory-like phenotype. In obese conditions, not only ATMs, but also macrophages from other tissues become metabolically active as will be discussed below.
Obesity affects macrophage metabolismWith regards to the macrophage’s metabolic state, two general phenotypes have been defined in adipose tissue. Macrophages termed redox-regulatory (Mox) are found in lean subjects, and metabolically activated macrophages (MMe) are found in obese adipose tissue [20]. In this section we will highlight metabolic states of general adipose tissue MMes, and of other subpopulations of macrophages residing in other microenvironments (Figure 2).
Lipid metabolism: In obesity, fatty acid composition of adipocytes is derived from dietary fatty acids. The same is observed for macrophages, but these cells also acquire fatty acids from dying adipocytes of hypoxic areas of the obese adipose tissue [21]. In obesity, the consumption of ω-6-polyunsaturated fatty acids such as arachidonic acid and linoleic acid, induces the machinery in adipocytes for synthesising inflammatory eicosanoids [22]. Leukotriene B4 (LTB4) is one of these eicosanoids, and has a central role in the recruitment of immune cells, and insulin resistance associated to systemic inflammation [23]. Free fatty acids (FFAs) released by dying adipocytes are engulfed by macrophages and participate in the activation of MMe [21]. These cells are involved in both detrimental and beneficial actions during obesity. Studies have observed that ATMs in obesity can overexpress both pro- or anti-inflammatory markers via signaling pathways that cannot be directly associated with cell surface markers of classical activation.
A study identified that ATMs from diet-induced obese mice or humans show weak expression of surface markers associated with pro- and anti-inflammatory states, but high expression of proteins linked to lipid metabolism such as ABCA1 (ATP binding cassette subfamily A member 1), CD36, and PLIN2 (perilipin 2). When human monocytes-derived macrophages or murine bone marrow-derived macrophages are stimulated with metabolic substrates (glucose, insulin and palmitate), the expression of metabolic markers is dependent on peroxisome proliferator-activated receptor (PPAR)-γ [24], a transcription factor which regulates lipid metabolism and promotes anti-inflammatory macrophage responses [25]. Interestingly, TNF-α (tumor necrosis factor alpha) and IL(interleukin)-1β expression is down-regulated by PPARγ [24], suggesting that PPARγ attenuates pro-inflammatory cytokine production during metabolic activation in ATMs of obese subjects (Figure 2).
In another study, ATMs of leptin-deficient obese animals (ob/ob) express a transcriptional program of lysosome biogenesis, and lysosome bodies are highly detected in this population. The ATMs do not alter the expression of pro-inflammatory genes, but CD206 (MRC1) is higher expressed in ATMs from obese mice compared to lean. These ATMs also present high amounts of lipid droplets accumulated in the lysosomes. In vitro, the differentiation of bone marrow-derived macrophages in the presence of digested adipose tissue increases the expression of Arg1, Il1b, and Nos2, genes involved in lipid uptake (Msr1 and Plin2) and lysosome genes (Atp6v0d2, Lipa, and Ctsk). In these cells, lipolysis is dependent on the activation of the liposome program, independent of their inflammatory phenotype [26] (Figure 2).
In other types of macrophages (Raw264.7, bone marrow-derived, or thioglycolate-elicited), the metabolic sensor adenosine monophosphate (AMP) kinase (AMPK) promotes a key signaling pathway that control macrophage lipid metabolism [27]. In a metabolic dysregulated system, AMPK antagonizes biosynthetic pathways and promotes catabolic processes through regulation of total mitochondrial content mediated by the activation of peroxisome proliferator-activated receptor gamma coactivated 1-(PGC1)α [28]. Mice fed a high fat diet (HFD) that do not express AMPK enhance ATM inflammation and insulin resistance [29] (Figure 2). In this sense, it is suggested that fatty acid oxidation (FAO) in ATM may protect obesity-associated complications while a fatty acid synthesis program in macrophages worsens obese adipose tissue complications [30].
Not only ATMs show distinct activation states in obesity. Macrophages from other microenvironments can also be activated distinctively [31,32]. Recently, it was shown that peritoneal macrophages from obese and diabetic mice have an anti-inflammatory phenotype that may explain why obese individuals have an impaired immune response against pathogens, whilst having a predisposition to develop tumors. In this study, peritoneal macrophages display a high expression of liver X receptor (LXR) and sterol regulatory element-binding protein (SREBP) targets, such as Idol, Srebf1, Srebf2, Scd1, and Abca1 (Figure 2). This study did not explore mechanisms for the expression of such metabolic markers, but it was demonstrated that weight loss restores peritoneal macrophage function and thus contributes to the reduction of immune-related comorbidities in patients [33]. The studies in lipid metabolism of macrophages in obesity have found similar results stating that obesity promotes a metabolic program in macrophages associated with upregulation of an anti-inflammatory state [24,26,29,33].
Glucose metabolism: A different study in mice and humans showed that in obese ATM fatty acid oxidation, glycolysis and glutaminolysis contribute to cytokine release by ATMs, but glycolysis seems to be the most important pathway for inflammatory cytokine production [34] (Figure 2). Indeed, it was shown that ENOblock, an inhibitor of pyruvate synthesis, increases the expression of anti-inflammatory markers in ATMs [35]. ENOblock-treated mice also have lower LDL/VLDL cholesterol in circulating blood, declined serum level of free fatty acids and less expression of inflammatory markers in stimulated Raw264.7 macrophages [36]. Classically, active HIF-1α promotes glycolysis by inducing the expression of enzymes in the glycolysis pathway (Figure 2). However, HIF-1α appears to play no critical role in pro-inflammatory activation of ATMs during early stages of obesity [37]. Conversely, macrophage HIF-2α ameliorates adipose tissue inflammation and insulin resistance in obesity [37].
Regarding the pentose phosphate pathway (PPP), this pathway is associated with adipose tissue inflammation and systemic insulin resistance in obesity. Mira Ham et al. reported that in obese ATMs, G6PD expression is higher in both humans and mice with obesity [38]. Moreover, G6PD is upregulated by FFA in primary macrophages isolated from the peritoneum of obese animals and induces the production of pro-inflammatory cytokines [39] (Figure 2).
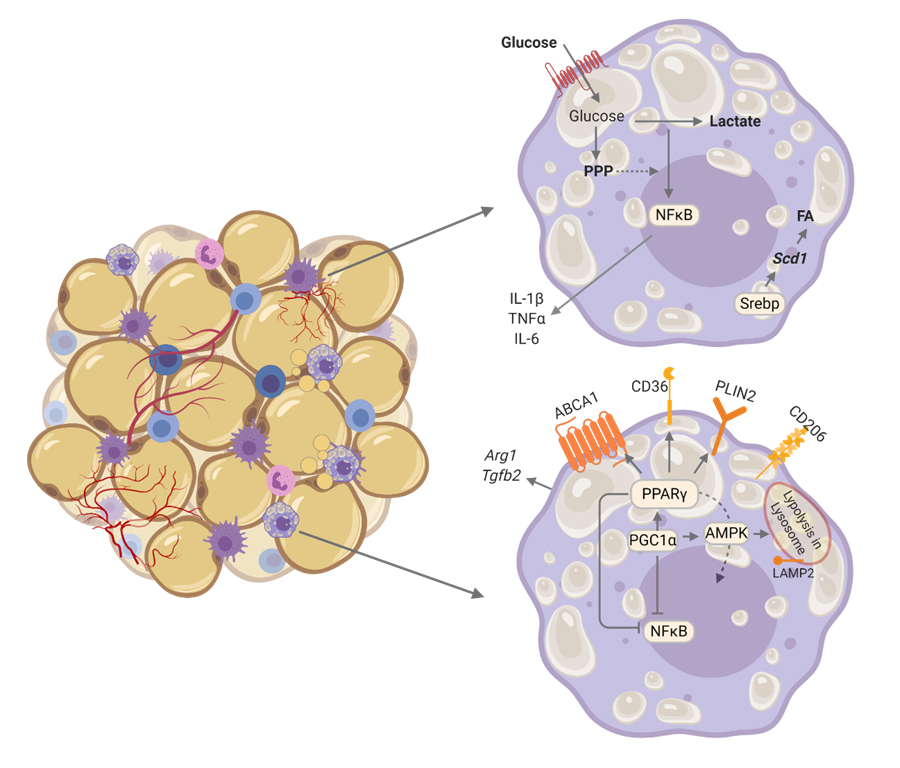 Figure 2. Metabolic pattern of adipose tissue macrophages (ATMs) in obesity. Studies have reported upregulation of glycolysis which is associated with higher expression of inflammatory cytokines in ATMs. In other studies ATMs also have upregulation of lipid metabolism pathways engaged by AMPK, PGC1α, PPARγ and lysosomes. This pattern upregulates genes involved in regulating lipid trafficking such as ABCA1, CD36, PLIN2, CD206, Arg1 and Tgfb2 (associated with pro-resolutive responses), while downregulates inflammatory genes such as IL1b, Tnf and Il6. The distinct findings among studies may be due to collection of different populations of ATMs, or even due to different timing among the studies in ATMs.
Figure 2. Metabolic pattern of adipose tissue macrophages (ATMs) in obesity. Studies have reported upregulation of glycolysis which is associated with higher expression of inflammatory cytokines in ATMs. In other studies ATMs also have upregulation of lipid metabolism pathways engaged by AMPK, PGC1α, PPARγ and lysosomes. This pattern upregulates genes involved in regulating lipid trafficking such as ABCA1, CD36, PLIN2, CD206, Arg1 and Tgfb2 (associated with pro-resolutive responses), while downregulates inflammatory genes such as IL1b, Tnf and Il6. The distinct findings among studies may be due to collection of different populations of ATMs, or even due to different timing among the studies in ATMs.
As mentioned in this section, altered metabolic patterns can be found distinctively in different subpopulations of macrophages in obese individuals [26,30,33–39]. The same can be observed for pro and anti-inflammatory markers [26,30,33–39], and in some cases the metabolic state does not depend on the inflammatory phenotype. In the previous section, we mentioned that adipose tissue has different subpopulations of macrophages (Figure 1). Possibly these subsets and localization were not identified in the studies mentioned in this section, which explains the different findings in distinct patterns of activation among the studies regarding glucose and lipid metabolism. Even different timings of animal sacrifice and macrophage collection can bring distinct patterns of activation. The activation states of macrophages can be triggered by excessive availability of nutrients and obesogenic factors that enable macrophages to acquire a pro-tumoral state which could benefit tumor development [40,41]. This will be further discussed after the next section in which metabolism of TAM will be explored.
Tumors exhibit a repertoire of recruited normal cells that contribute to the TME through promotion of hallmark traits [42]. In solid tumors, TAMs can derive from both, tissue-resident-macrophages and circulating monocytes, and this ontogeny may define TAMs function in tumor progression [43]. In perivascular or hypoxic areas, TAMs display proangiogenic and immunosuppressive effects [44], while TAMs in the invasive tumor front or cancer cell nests promote anti-tumoral activity [45,46]. But these functions associated with TME localization may vary according to the type of tumor, age, nutritional condition, etc. In simplified classifications of macrophages, pro-inflammatory macrophages are associated with tumoricidal activities. But the pro-inflammatory phenotype can contribute to cancer initiation by producing pro-inflammatory mediators such as IL-6, TNFα and IFNγ, growth factors including VEGF and WNT, proteases, reactive oxygen species (ROS) and reactive nitrogen species (RNS), which together create a mutagenic TME [47,48]. Macrophages can induce adaptive immune responses through the presentation of tumor antigens and the production of chemokines and cytokines to attract and activate CD8+ effector T cells and natural killer (NK) cells. Moreover, macrophages can promote direct cytolysis of cancer cells through antibody-dependent cellular cytotoxicity, which involves recognition and binding of an antibody-coated cancer cell by the macrophage [49].
As the tumor grows, recruitment of macrophages to hypoxic areas, and Th2 cell-derived IL-4 can reprogram macrophages to become pro-tumoral [50,51]. Th2 cells can migrate to the TME after antigen-loaded DCs prime them in draining lymph nodes. In the TME, attracting chemokines are released by dendritic cells (DCs) which are activated by cancer-associated fibroblasts [51]. A high ratio of Th2/Th1 in the TME is associated with less survival [52]. As the TME is modified, tumor progression is supported by suppressing immune cell-mediated cytotoxicity [53]. TAM-induced immune suppression is mediated by the expression of inhibitory receptors and checkpoint ligands, such as PDL-1 and PDL-2, which inhibit NK and T cell functions [54]. TAMs also secrete several cytokines, such as IL-10 and TGFβ, that contribute to the immunosuppressive microenvironment by inhibiting Th1 cells, and CD8+ T cells and by inducing regulatory T (Treg) cell expansion. Tumor progression requires an “angiogenic switch” characterized by new vessels through recruitment of perivascular cells [55]. TAMs can support angiogenesis by the production of vascular endothelial growth factor (VEGFA) and pro-angiogenic chemokines CXCL1, CXCL8, CXCL12, and TGFβ [56]. Metastasis can also be influenced by macrophages though promotion of extravasation, survival and growth signals to tumor cells and inhibition of cytotoxic T cells [57]. These factors also promote the recovery of tumors from biologic therapies, chemotherapies and radiotherapies. In some tumors, the neutralization of reactive CD8+ T cells by macrophage infiltration also interferes with efficacy of immunotherapies [58].
As previously mentioned, obesity is a multifactorial condition in which subpopulations of macrophages acquire distinct activation states, including ones that show a less inflammatory state which relies on a dysregulated metabolism. In obesity, besides macrophages, NK and CD8+ T cells have a similar phenotype which impairs tumoricidal responses [59,60]. Therefore, dysregulated metabolism in immune cells contributes to strengthen the TME and worsens the quality of life of obesity-associated cancer patients.
The Metabolism of Tumor-Associated MacrophagesThe metabolic profile of TAMs is very dynamic, and the nutritional need of malignant cells is one of the factors that affect TAMs metabolism. Not only TME affects TAMs, but TAMs also modulate metabolic pattern of the whole TME [61]. In a recent clinical and pre-clinical study it was demonstrated that TAMs enhance tumor hypoxia and glycolysis of subcutaneous tumors in mice and patients with non–small cell lung cancer (NSCLC) [61]. In this study, TNF-α secreted by TAMs promotes glycolysis in tumor cells, whereas increased AMPK and PGC1α in TAMs facilitate tumor hypoxia (Figure 3). Depletion of TAMs by clodronate abrogated aerobic glycolysis and tumor hypoxia, improving tumor response to anticancer therapies, whilst increasing PD-L1 expression in cancer cells which consequently improved antitumor efficacy by PD-L1 antibodies as well as T-cell infiltration in tumors [61]. The metabolic interactions in the TME have emerged as a hallmark of cancer [42], where in this scenario, the nutritional status of the individual may interfere in the composition of the substrates that feed TAMs and the TME. Next, we will discuss how the metabolism of different substrates on TAMs may influence tumor progression and how this can be amplified by obesity. (Figure 3).
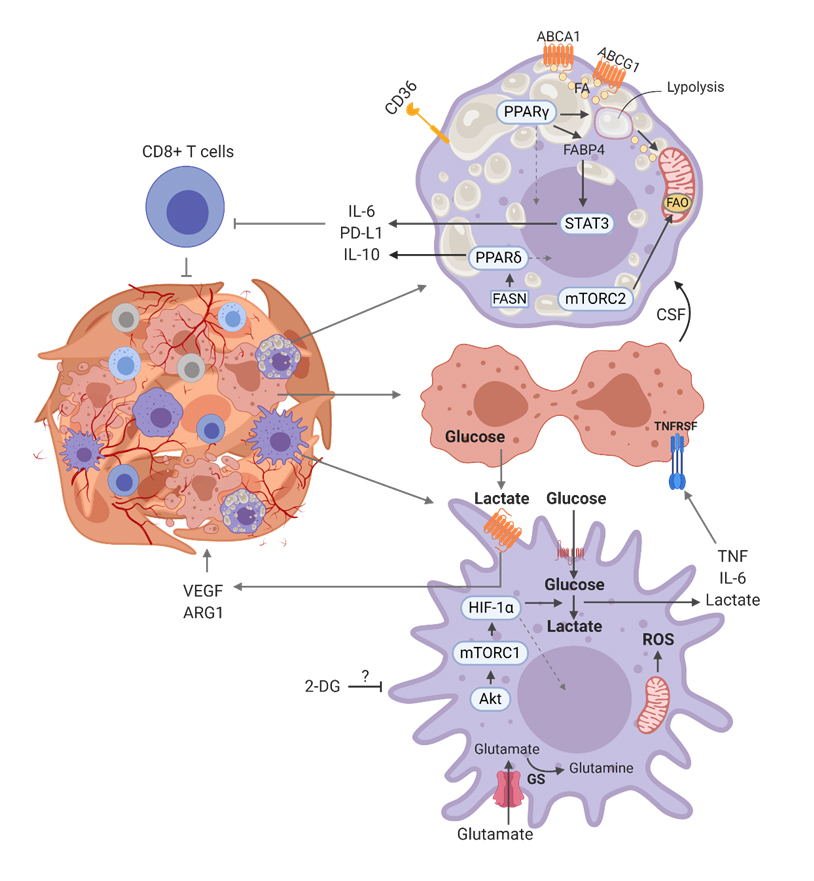 Figure 3. Metabolic pattern of general tumor associated macrophage. Depending on the type of cancer, literature has mentioned macrophage glycolysis as a pro-tumoral pathway [62–65], that promotes tumor Warburg effect in tumor cells in a TNFα dependent manner. Lactate derived from glycolytic tumor cells promotes secretion of VEGF in macrophages which contributes to angiogenesis in the TME. Lipid catabolism and lipid efflux have been associated with pro-tumoral activity due to the promotion of immune pathways that deactivates cytotoxic CD8+ T cells.
Figure 3. Metabolic pattern of general tumor associated macrophage. Depending on the type of cancer, literature has mentioned macrophage glycolysis as a pro-tumoral pathway [62–65], that promotes tumor Warburg effect in tumor cells in a TNFα dependent manner. Lactate derived from glycolytic tumor cells promotes secretion of VEGF in macrophages which contributes to angiogenesis in the TME. Lipid catabolism and lipid efflux have been associated with pro-tumoral activity due to the promotion of immune pathways that deactivates cytotoxic CD8+ T cells.
Glucose metabolism: In the TME, TAMs compete with cancer cells for nutrients, particularly glucose, and therefore undergo changes in their glucose metabolism, in a similar manner as the tumor cells do [66] (Figure 3). Hypoxic areas of the TME promote a selection pressure, with the most aggressive cells surviving these hostile growth conditions [67]. A study showed that hypoxia in the TME inhibits mTORC1 in TAMs, shifting their metabolism towards less glucose uptake, increasing glucose availability in the TME and promoting hyperactivation of endothelial cells, resulting in improvement of angiogenesis [68]. Colegio et al. demonstrated that tumor-derived lactate promotes HIF-1α stabilization, and transcription of ARG1 and VEGF in TAMs [68,69].
In normoxia, glycolysis in TAMs stimulates the progression of the TME. Lactate secretion by human carcinoma cells causes a switch from oxidative phosphorylation to glycolysis in TAMs which is accompanied by increased lactate, TNF-α, and IL-6 production [62]. Similarly, when human monocytes are stimulated with thyroid carcinoma-conditioned medium, they activate AKT1/mTORC1 pathway [63]. Corroborating this, there is increased expression of HK2 in TAMs from MMTVPyMT mice with tumor extract solution from a breast cancer patient [63]. Indeed, the Warburg effect in human monocytes co-cultured with pancreatic ductal adenocarcinoma (PDCA) cell lines promotes vascular network formation, increased extravasation of tumor cells out of blood vessels and induces epithelial-to-mesenchymal (ETM) transition [64]. In clinical studies, 2-DG is being tested in cancer treatment associated with radiotherapy [70–72]. In summary, these studies suggest that glycolysis in tumor cells or in TAMs can contribute to cancer progression. In obesity, elevated glucose levels has a central role in the development of obesity-associated complications, such as insulin resistance which also improves tumor progression [73]. Thus, high availability of glucose in obesity can enhance tumor growth through promotion of the Warburg effect in tumor cells in TAM-induced hypoxic areas, as well as glucose uptake and glycolytic pathway upregulation in TAMs residing in normoxic areas.
Lipid metabolism: Besides glucose metabolism, TAMs also change their lipid metabolism, including FA uptake, biosynthesis, storage, and oxidative pathway (Figure 3). In breast cancer, apoptotic tumor cells release miR-375 which binds oxidized phospholipids and low-chain fatty acids to be taken up by TAMs via CD36. This enhances macrophage migration through CCL2 and promotes tumor progression [74]. Regarding lipolysis and cholesterol efflux, the downregulation of medium-chain acyl-CoA dehydrogenase (MCAD) in TAMs, by deleting the PPARγ pathway, reduces lipid catabolism and inhibits tumor growth. In another study, the lack of monoacylglycerol lipase (MGLL) leads to lipid overloading in TAMs, which in turn affects tumor progression. In another study, it was observed that hyaluronic acid produced by cancer cells promotes cholesterol efflux in TAMs and consequent tumor progression [75]. These examples lead us to speculate if lipid accumulation would improve anti-tumoral responses in TAMs, while lipolysis and cholesterol efflux in TAMs would promote tumor growth. In obesity, NK cells reprogram their metabolism towards PPARδ-driven lipid accumulation, preventing the activity of the cytotoxic machinery to the NK cell–tumor synapse [59]. Similarly, cholesterol inhibits IL-9 secretion in CD8+ T cells by activating LXR pathway, and this impairs cytotoxic responses in lymphocytes [60]. In previous sections, we discussed similar phenotype in macrophages of obese subjects [33]. Based on these examples, it is suggestive that dysregulated lipid accumulation and its consequent inhibition of anti-tumor responses is conserved among different immune cells in obesity.
Regarding fatty acid metabolism, it was shown that pharmacological FAO inhibition favors an antitumorigenic polarization in TAMs from colorectal cancer [76]. Indeed, FAO has important effects on the induction of immunosuppressive TAMs. Recently, Wu et al. observed that lipid droplet-dependent FAO triggers an immunosuppressive TAM phenotype through mTORC2 [77]. Yet in myeloid cells, fatty acid synthase (FASN) is upregulated as a consequence of colony stimulating factor 1 (CSF1), secreted by lung cancer cells [78]. Interestingly, IL-10 secretion is dependent on PPARδ, however PPARδ activation is blocked when FASN is knocked down in myeloid cells, implying that endogenous ligands derived from FASN in myeloid cells can be a key regulator of PPARδ activity in the context of tumor myeloid cells [78]. In recent studies, TAMs infiltrating early human breast cancers display high expression of fatty-acid binding protein (FABP)5, which associates with lipid droplet accumulation. At this stage, TAMs present an anti-tumoral phenotype through secretion of IFN-I. In the late stages, infiltrating macrophages express FABP4, which supports tumor progression by favoring IL6/STAT3 signaling [79].
Lipid signaling has also been associated with tumor progression. The expression of 5-lipoxygenase (5-LO) in hypoxic areas of the ovarian TME is correlated with strong infiltration of TAMs. 5-LO metabolites such as LTB4 are shown to promote migration and invasion of macrophages, mediated by the upregulation of matrix metalloproteinase (MMP)-7 expression [80]. Migration is classically attributed to 5-LO products in several inflammatory contexts, such as in hypertrophic adipose tissue of obese subjects [23]. Since LTB4 is detected and exerts pro-inflammatory functions systemically in metabolic syndromes [23,81], LTB4 in cancer can be one of the mechanisms by which obese subjects have a poor prognosis of cancer. Additionally, TAMs isolated from human renal cell carcinomas show increased activity of 15-LO, and increased secretion of 15(S)-HETE. The production of CCL2 and IL-10 is also increased through a 15-LO-dependent mechanism [82], suggesting lipoxygenases products as potential inducers of tumor growth.
Most of the studies reported in this topic are in tumor models that are currently associated with obesity, such as breast [74,79], colorectal [76], renal [82], and ovarian cancer [80]. Since obesity is featured by hyperlipidemia and different pathways of the lipid metabolism induces an immunosuppressive phenotype [83], it is suggestive that the high availability of lipids in an obese condition would enhance such pathways in TAM and subsequent tumor progression.
Amino acid metabolism: Beside glucose and lipid metabolism, TAMs can also show dysregulated amino acid metabolism. As such, TAMs express high levels of glutamine transporters [69]. Accordingly, pharmacological inhibition of glutamine synthetase (GS) skews pro-tumoral macrophages toward the anti-tumoral phenotype, characterized by increased succinate with enhanced glycolysis, which is partly related to HIF1α activation. GS-inhibited macrophages also induce T cell recruitment, reduced T cell suppressive potential, and ability to foster angiogenesis and metastasis [84] (Figure 3). Altered arginine metabolism can also result in pro-tumoral TAM functions and is a consequence of hypoxia- and/or lactate-mediated induction of Arg1 [69]. Macrophages deficient in Arg1 develop significantly smaller tumors than the wild-type mice. Interestingly, this phenotype in TAMs is mediated by hypoxia inducible factor (HIF-1α), a pathway often observed in glycolytic macrophages [69]. Regarding tryptophan metabolism, it is important to mention that the enzyme IDO (indoleamine 2,3-dioxygenase 1) expressed in TAMs can culminate in a functional impairment of T cells in the TME, as shown in a study in which TAM-derived IL-12 is required for early T cell activation and subsequent IDO expression in TAMs. Conditioned medium from IDO(+) TAMs effectively suppressed T cell responses in vitro [85]. These findings suggest that the metabolism of amino acids may play an important role in the function of TAMs. It has been reported that the expression of IDO and ARG1 in macrophages is highly pronounced in obese individuals [86–88]. Moreover, obese individuals have altered glutamine metabolism [89]. The metabolism of the three amino acids in TAMs seems important for tumor growth, and obesity may worsen the dysregulated metabolism of such amino acids which would potentiate the TME.
In any cell, metabolic pathways are integrated, where one pathway can affect the flow and machinery of another. This is observed in TAMs when lactate from tumor cells provokes a switch from FAO to glycolysis or vice-versa [61,68]. This may vary with time-space of TAMs in the TME. In obesity, such pathways are dysregulated systemically in adipose tissue and in immune cells, such as macrophages of different microenvironments. This is promoted by several obesogenic factors that can affect directly (or indirectly) tumor development and progression. These obesogenic factors and their effects on amplification of TAMs’ signal in promoting tumor progression will be explored in the next section.
Reports from WHO and World Obesity Federation have declared obesity as an epidemy [10]. Many conventional non-metabolic diseases are associated with obesity, such as some types of cancer. Indeed, up to 49% of certain types of cancer are now attributed to obesity [90]. Prevalent cancers in obese individuals are mainly endometrial, esophageal adenocarcinoma, colorectal, postmenopausal breast, prostate and renal [91,92]. Although male breast cancer (MBC) accounts for 1% of all breast cancer, MBC incidence has increased in the last few decades, and different studies have attributed this increase to the exponential rise in the number of overweighed and obese people [93]. Obesity can worsen distinct aspects of cancer, including quality of life, cancer recurrence and progression, and patient survival [94,95].
In obesity, hyperadiposity leads to metabolic disorders, altered production of hormones and chronic systemic inflammation [96–98]. These pathophysiologic effects have been associated with tumor development and progression [96,98,99]. This association has been supported by animal experiments in which obesity and cancer have been modified by dietary means [100]. Different studies have highlighted that in an overnutrition context, lipid-laden immune cells such as NK cells and CD8+ T cells have their anti-tumoral responses affected negatively [59,60]. Recently, similar results were observed in macrophages, in which lipid metabolism in TAMs elicits an immunosuppressive phenotype [77]. In fact, hepatocellular carcinoma is accelerated by steatosis involving immunosuppressive markers in macrophages mediated by HIF-1α [101].
The mechanisms linking obesity to cancer have shown up within the past decade. In both situations, excessive nutrients promote a distinct immunosuppressive state. Due to macrophages composing a huge portion of immune cells that infiltrate in tumor and adipose tissue, these cells are possible candidates to support tumor development and progression in the context of metabolic diseases, because of the similar effects that macrophages promote in both situations. Different factors may link obesity to pro-tumoral TAMs as discussed above (Figure 4).
Obesity Factors Improve TAM-Promoted TMESeveral factors can directly link obesity to tumor growth when macrophage modulation is the main issue as represented in Figure 4. Fatty acid composition of TAMs can be modulated by dietary fatty acids [22]. It is known that ω-3-PUFAs can neutralize the inflammatory effects of ω-6-PUFA in macrophages [22]. In tumor cells, ω-3-PUFAs reduces cancer cell proliferation, development and progression [102], suggesting that in obesity, inflammatory PUFAs can potentiate the development of the TME through activation of TAMs, while ω-3 would interfere with the TME by downregulating TAM activation. This suggests that dietary intervention with ω-3-PUFAs can prevent cancer development in obese subjects or can diminish the complications of obesity-associated cancer patients.
In obese adipose tissue, hypoxic areas promote a favorable environment for tumor cells. Indeed, the development of breast cancer cells is linked to hypoxia [103]. As TAMs contribute to hypoxia in TME [61], the poor prognosis of breast cancer in obese subjects is possibly linked with higher frequency of hypoxic areas promoted by TAMs. In obesity, macrophages from different microenvironments acquire a phenotype in which the machinery of collagen and ECM secretion is activated [104]. Springer et al observed that obesity-associated interstitial fibrosis promotes a macrophage phenotype similar to tumor-associated macrophages [105]. Therefore, such a chronic state would improve the tumorigenic phenotype in ATMs which would promote adipose tissue associated tumors, such as breast cancer. Elevated levels of leptin is found in the circulation of obese individuals [106]. Also, leptin has been reported to be associated with an enhanced risk for different types of cancer such as colorectal, hepatocellular, renal, breast, ovarian, endometrial and prostate [107]. In fact, leptin induces the secretion of IL-18 by TAMs through activation of NFκB signaling, which contributes to migration and invasion of breast cancer cells [108]. In another study, leptin promot tumor growth and metastasis by stimulating IL-8 production through activation of p38 and ERK1/2 in TAM [109].
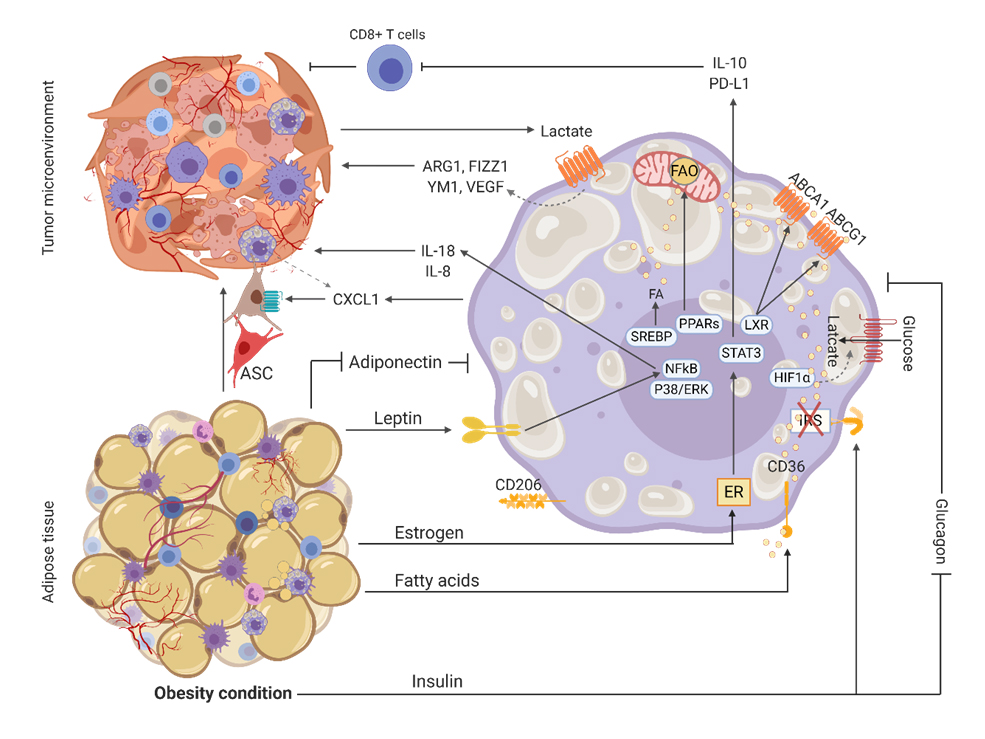 Figure 4. Obesity factors that support tumor microenvironment through modulation of tumor-associated macrophages. Leptin acts on its receptor in TAMs resulting in the secretion of inflammatory factors that are important for the progression of TME. Yet estrogen and insulin provide an immunosuppressive state that impairs the activation of cytotoxic lymphocytes. Adipose stromal cells (ASC) can leave adipose tissue and be recruited by CXCL1 released by TAMs in the TME. Fatty acids (FA) can be synthetized or taken up from lipolysis in TAMs or tumor cells. In TAMs, FA can follow the efflux pathway which culminate in the immunosuppressive state or they can overload and inhibit mitochondrial respiration, culminating in upregulation of glycolysis. The Warburg effect in tumor cells releases lactate as a byproduct which activates the transcription of markers involved in extracellular matrix organization and angiogenesis.
Figure 4. Obesity factors that support tumor microenvironment through modulation of tumor-associated macrophages. Leptin acts on its receptor in TAMs resulting in the secretion of inflammatory factors that are important for the progression of TME. Yet estrogen and insulin provide an immunosuppressive state that impairs the activation of cytotoxic lymphocytes. Adipose stromal cells (ASC) can leave adipose tissue and be recruited by CXCL1 released by TAMs in the TME. Fatty acids (FA) can be synthetized or taken up from lipolysis in TAMs or tumor cells. In TAMs, FA can follow the efflux pathway which culminate in the immunosuppressive state or they can overload and inhibit mitochondrial respiration, culminating in upregulation of glycolysis. The Warburg effect in tumor cells releases lactate as a byproduct which activates the transcription of markers involved in extracellular matrix organization and angiogenesis.
In obesity, high levels of insulin can be found in circulation as a compensatory effect of peripheric insulin resistance. This is a well-established risk factor for cancer [73,110] which is linked with systemic inflammation [111]. A recent work identified that insulin-resistant macrophages possess an M2-like phenotype, reduced LPS responses and exhibit increased glycolysis [112]. These results suggest that beside the direct effects of insulin in tumorigenesis [113], insulin would also act directly in macrophages to promote an insulin dependent pro-tumoral state. Glucagon opposes the actions of insulin by mobilizing glucose and inhibiting its utilization. Analogues of glucagon are used to suppress the invasion of pro-inflammatory macrophages into adipose tissue [114]. They are also used to inhibit cancer development because they induce cell apoptosis in some tumors in some cases by inhibiting glycogen synthase kinase-3 (GSK-3) [115]. This suggests that analogues of glucagon could be used as a complementary therapy to prevent cancer in metabolic diseases. Likewise, adiponectin levels are lower in obese people in comparison to individuals in the regular BMI range [116]. There is a negative association between circulating adiponectin levels with cancer risk and disease severity [117]. It has been reported that the hormone inhibits VEGF via AMPK activation [118], and consequently inhibits angiogenesis in cancer. In macrophages, adiponectin induces the secretion of pro-inflammatory cytokines [119], suggesting that, depending on the stage, adiponectin would promote anti-tumor properties in TAMs. Further factors influencing breast cancer development via obesity include estrogens produced in adipose tissue. Estrogen can also be stimulated by oral contraceptives, hormone replacement therapy, and pregnancies [120]. Leptin increases pro-estrogenic pathways, estradiol production and estrogen receptor-α signaling [121], all of which have particular significance in estrogen-responsive cancers. In macrophages, estrogen stimulates a pro-resolutive phenotype, suggesting that in excessive amounts such as in obesity, estrogen would add to breast carcinogenic state in macrophages.
Another aspect that composes the development of cancer in obese subjects involves several studies showing that adipose stromal cells (ASC) infiltrate tumors, and not other organs, and contribute to the pro-tumoral microenvironment by facilitating tumor vascularization [122]. This is enhanced in obese subjects, and in this condition ASCs migrate to the tumor in response to tumor-derived CXCL1 [123]. It is not known which cell population produces CXCL1, but in parallel, a study showed that CXCL1 is the most abundant chemokine secreted by TAMs, and CXCL1 can promote breast cancer migration and invasion ability [124]. Similarly, studies found that ASCs promote metastasis and alterations in the extracellular matrix mechanics when they invade breast TME [125], suggesting that in obesity TAMs would partially contribute to TME and invasion though recruitment of ASCs into the tumor.
Glucose and lipids are both substrates abundantly circulating in obese subjects. In abundance, both substrates can feed the TME and feed tumor cells and non-tumor cells, such as TAMs, exacerbating the pathways that induce such TME conditions. In fact, obesity-associated lipolysis prompts the release of fatty acids. Accumulation of fatty acids in turn affects proper responses in macrophages, forming a vicious cycle. Indeed, a research group demonstrated that the ATMs promote triple-negative breast cancer occurrence in obesity by releasing IL-6, which can bind GP130 (glycoprotein 130) on tumor cells and induce stem-like properties. This effect could be reversed by losing weight [126].
As previously discussed, the systemic metabolic context of a subject with cancer may reflect the metabolic state of the TME, and possibly this may be the cause for a limited success in tumor therapy that includes immunotherapy. Thus, personalized therapies based on the context of systemic versus TME metabolic state would be relevant in order to direct an individual therapy to try a better success rate. Controlling circulating factors (glucose, lipids, hormones) that promote comorbidities associated with obesity and alter macrophage function, would possibly impact tumor development, since these cells have phenotypic plasticity as one of their main features. Willemsen et al., 2019, observed in diet-induced obese mice that weight loss reversed the deactivated macrophage phenotype [33]. Such deactivated phenotype may reflect pro-tumoral features of TAMs that would be reversed in a context in which obesity comorbidity-associated factors are well-controlled.
Another issue raised by obesity promoting TAMs, is the clinical utilization of drugs that target cell metabolism. These drugs are not cell specific, and depending on the situation, unspecific anti-metabolic therapies can worsen the clinical condition mainly in subjects with obesity [127,128]. Therefore, individual cell specific therapies would work better. Although ATM and TAM metabolism have similarities, targeting TAM metabolism would be more effective. As mentioned in subsection Obesity affect ATM in subtissular niches, some subtypes of ATMs, such as VAMs, LAMs and CD11b+Ly6c+, may mitigate inflammatory dysregulation in hypertrophic adipose tissue due to their effects on angiogenesis and adipogenesis [16,17,19]. The inhibition of those subtypes would worsen the hypertrophic adipose tissue complications since it would promote more hypoxia and enlarged adipocytes. While the improvement of those subtypes’ functions would favor tumor expansion, mainly those located in or near the adipose tissue. Thus, we suggest that targeting ATMs would worsen more than mitigate obesity complications that contribute to the TME.
A way to target TAM metabolism specifically would be through the use of nanoscale drug-delivery systems. Such technology has been experimentally developed for conventional anti-tumoral drugs targeting tumor cells [129,130], but the cytotoxicity of such drugs can worsen the clinical condition due to the proliferation signals that cell death provoke in TME [42]. Alternatively, the target could be tumor antigen-loaded macrophages, and less toxic inhibitors of metabolic pathways, such as PPAR-γ ligands [131], could be added to nanoparticles to specifically target TAMs metabolism in attempt to alter their pro-tumoral functions. Evidently, the mitigation of any drug toxicity, off-targets and nanoscale drug-delivery systems must be better explored. Also, any therapy would be more effective if individual metabolic conditions are well described. Thus more knowledge about macrophage metabolism in different types of cancers and metabolic conditions must be urgently explored in order to suggest which metabolic pathways can be targeted to neutralize pro-tumoral properties in macrophages in specific conditions. Therefore, while there is no magic pill to prevent and ameliorate cancer, obesity and both pathologies together, nutritional and physical activity education are currently quite relevant interventions to improve quality of life, reduce risk of cancer and improve outcomes.
JVdB designed the outline of the review and supervised during the writing process, SY and TR wrote the paper, SY made the figures and all authors proofread the text before submission.
The authors declare that they have no conflicts of interest.
JVdB received a VENI grant from ZonMW (91615052), a Netherlands Heart Foundation Junior Postdoctoral grant (2013T003), and Senior Fellowship (2017T048), a NWO ENW-KLEIN-1 grant (268) and a CCA PhD grant from Cancer Center Amsterdam.
We thank Karl Harber for proofreading the manuscript as a native English speaker.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
117.
118.
119.
120.
121.
122.
123.
124.
125.
126.
127.
128.
129.
130.
131.
Yang S, Van den Bossche J, Ramalho T. Macrophage Metabolism at the Crossroad of Metabolic Diseases and Cancer. Immunometabolism. 2020;2(3):e200022. https://doi.org/10.20900/immunometab20200022

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




